碳纳米管限域的金属纳米粒子的催化行为
- 格式:doc
- 大小:61.00 KB
- 文档页数:6

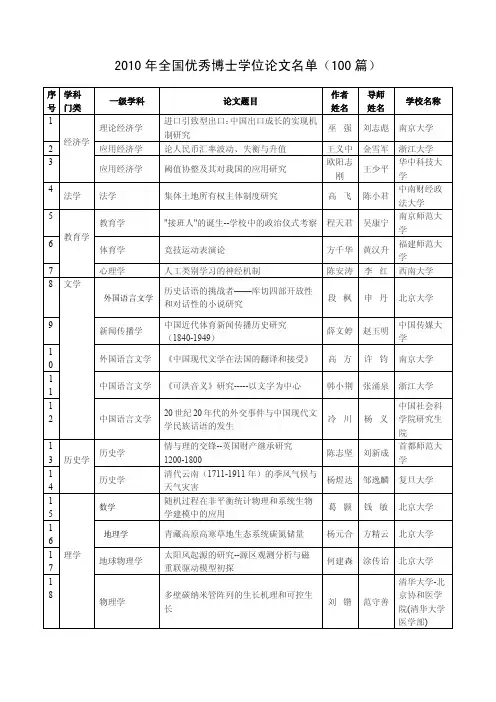


中国科学: 化学 2012年 第42卷 第4期: 355 ~ 362SCIENTIA SINICA Chimica 《中国科学》杂志社SCIENCE CHINA PRESS评 述中国科学院学部 科学与技术前沿论坛 催化中国专刊催化基础理论研究发展浅析——兼述催化中的限域效应 (代序)包信和*中国科学院大连化学物理研究所, 催化基础国家重点实验室, 大连116023 *通讯作者, E-mail: xhbao@收稿日期: 2012-03-12; 接受日期: 2012-03-15; 网络版发表日期: 2012-03-27 doi: 10.1360/032012-130摘要 催化的基础理论在过去的一个多世纪里已经有了长足的发展. 始于20世纪60年代的表面科学, 在为人们提供固体表面化学反应的分子图像以及反应与表面(电子)结构的关系上取得了重要进展. 得益于上世纪末发展起来的纳米科学和技术, 人们对催化的研究得以更加接近实际催化过程, 得到的结果有望为实现高效催化体系的合理设计提供基础. 本文简要回顾了催化基础理论研究发展的历程, 描述了当前纳米催化领域的研究前沿和挑战, 结合本实验室的研究结果, 着重分析和探讨了纳米催化中的限域效应.关键词 表面化学 纳米催化 限域1 引言催化过程, 是在化学反应过程中借助催化剂, 对化学反应进行选择、调控的化学过程. 作为关键和核心技术, 催化在合成氨工业、石油炼制、精细化学的合成、高分子材料的制备以及环境保护工程中起了非常重要的作用[1]. 自上世纪初以来, 催化技术经历了从合成氨到石油炼制、高聚物合成以及现在的酶催化和不对称催化等多次重大的技术突破, 产生了 F. Haber, J. Langmuir, K. Ziegler, G. Natta 和G. Ertl 等在内的十几位诺贝尔奖获得者. 但从本质上讲, 催化技术的创新还是工艺多于理论. 近代科学的发展为人类从本质上认识复杂的催化过程、从分子水平上对催化剂进行设计提供了可能性[1~4]. 随着公众对国民经济可持续发展和保护人类生存环境要求的日益提高, 与能源、环境、农业以及人类健康密切相关的化学工业正在经历着一场重大的革新, 作为主导和起关键 作用的催化科学, 也必将面临一场重大的技术革命.如果说迄今为止催化领域的研究重点在于寻找高效催化剂以提高生产力, 那么, 未来催化科学研究的主要任务是探索如何选择和控制化学反应, 使反应过程朝着人们所希望的方向展开. 新催化剂研制的中心目标是实现低温条件下的高转化率、接近100%的选择性和尽可能高的稳定性.早在1835年左右, “催化”这个概念就被提了出来. 经过近两百年的发展, 催化在理论和实践的诸多方面有了很大的发展, 但是催化相关的基本概念并没有发生根本性的变化. 现在看来, 真正上升为催化理论, 并且大家一致公认的不外乎下述几点: 一是Arrhenius 和Sabatier 在19世纪80年代提出的反应能垒理论; 二是Langmuir 在20世纪50年代建立的表面吸附理论; 再就是Taylor 等在20世纪50年代后提出的活性中心概念. 再后面发展出来的一些如 结构敏感性、择形催化和酸碱催化等概念都是在以上三点基础上衍生出来的. 这些理论和概念长期以来包信和: 催化基础理论研究发展浅析356为人们认识催化机理、理解催化过程和创制催化剂等提供了重要的理论基础和实践指南. 但是, 即使这些为数不多的概念和理论, 大多还仅仅是具有“定性”的意义, 往往多用来从表象上解释催化现象, 而真正意义上能够精确刻画催化过程, 从而指导催化剂设计的理论还非常少见. 甚至, 即便对已历经了一个世纪多的发展、先后催生出多名诺贝尔奖的氮加氢制备氨气的合成氨催化过程, 人们现在也很难说对它已经达到充分认识的程度[1, 5]. 随着经济社会的飞速发展和环境的急剧变化, 人们对催化科学和技术的发展提出了更高的要求: 催化基础要从传统的基于石油化工的碳-碳键活化拓展到面向煤转化和生物转化的碳-氢键和碳-氧键活化; 催化过程要探索从惯常采用的热激发过渡到先进的光、电等外场激发; 催化技术要从追求单一过程的高效转化发展到面向资源高效利用和产品多样化的选择性优先. 力求实现从分子水平上对化学反应过程的理解和调控, 并努力服务于原子经济的高效过程, 这是当今催化科学和技术发展面临的重要挑战. 为此, 就需要我们对催化剂和催化作用的本质有更深的认识, 在此基础上实现理论和概念的创新.2 催化中的价电子调控特性催化, 特别是多相催化的基础是反应物分子在催化剂表面的吸附和脱附. 反应物分子通过吸附在催化剂表面形成键合, 这种吸附分子与催化剂表面的成键(即使是弱相互作用), 导致了反应分子在结构和电子分布上发生畸变, 从而改变了反应过程的状态和途径. 这些变化的基础是吸附作用对反应涉及的活化能垒的调控, 而其本质是基于吸附分子与催化剂表面的电子传递(如图1所示). 我们将催化剂的表面作为一个“赝分子”, 根据“前线轨道理论”, 反应物分子跟催化剂表面这一“赝分子”的相互作用(成键)要遵循轨道的“对称性匹配”和“能量匹配”法则. 在不考虑轨道对称性的条件下, 这种成键的强弱决定于反应分子相关轨道与代表催化剂表面的“赝分子”轨道的能量匹配程度. 很显然, 改善和调控这种匹配关系可以有两条途径: 一是改变反应物的轨道分布, 使它能更好地与“赝分子”匹配, 实践中通常是采用热激发、光激发和其他可能的外场激发; 另一种是调变催化剂表面这个“赝分子”的轨道分布, 使它反过来与反应物分子更好地匹配, 从而实现对催化反应的调控.长期以来, 在类似概念的基础上发展出了一系 列催化调控的理论和方法. 早先, 人们认识到在催化剂中加入另外的元素组分(如图2所示), 能够很好地调变催化特性[1]. 最著名的例子是合成氨催化剂的发现和优化. 上世纪初, 德国BASF 公司以Mittach 等为代表的研究者, 在Haber 等早期发现熔铁催化剂(Fe 1 x O 和Fe 3O 4)的基础上, 经过大量的实验筛选, 在氧化铁中加入多种不同组分, 先后实验了两千多个配方, 最后终于成功获得了现今工业上还在广泛使用的含有近十种不同添加剂的氧化铁催化剂. 采用近代研究方法得到的结果表明, 这些添加剂的加入确实改变了氧化铁的价电子分布, 从而改变了作为图1 催化剂和催化过程中的电子传递和反应能垒示意图图2 催化剂电子特性的组分调控[6]中国科学: 化学 2012年 第42卷 第4期357该反应控速步骤的氮分子在催化剂表面的吸附和解离[5]. 通过添加不同组分调变催化性能的方法, 在实践中得到了广泛应用. 这种广泛的筛选往往被称之为“炒”催化剂, 实际上, 这种表面上的“炒”与催化作用有着内在的本质关联, 关键需要很好的理论指导和大量分析数据支撑.上世纪中叶以来, 随着表面科学和技术的发展, 人们发现, 材料表面, 即使是研究中广泛使用的规整单晶表面, 在原子层次上也不是平整的, 存在台阶、边角和缺陷. 在这些位置上, 由于所谓的原子配位数不一样, 导致了局域电子结构的不同. 这些发现使人们认识到除了调变体系的组分以外, 通过控制材料的表面结构, 如不同晶面、台阶和缺陷的分布等也能调变体系的电子结构[6, 7], 如图3所示. 这样, 人们又探索出了一条通过控制晶面和表面结构调控催化反应的途径. 这种基于规整表面和结构控制的研究历经整整半个世纪. 尽管得到的理论和实验结果大大丰富和提高了人们对催化基础的认识, 特别是对反应分子在表面成键和表面物理化学的认识, 代表人物之一, 德国Fritz-Haber 研究所的G. Ertl 教授也因此而获得2007年度诺贝尔化学奖, 但是并不像Mittach 他们那样幸运, 现在也很难说哪个催化剂的成功开发是完全基于这种表面结构调控理论和实践的. 以至于这一领域的另一个重要的代表人物, 美国加州大学伯克利分校的G. Somorjai, 也在近年对自己近50年的研究进行反思, 认为有些研究走了一定的弯路[8]. 现在看来, 基于规整表面的基础研究与实际催化剂作用存在较大偏差是低估了催化剂和催化作用的复杂性. 众所周知, 催化是一个在一定温度、压力和反应物分子存在情况下发生的动态过程. 已有的研究图3 催化剂电子特性的晶面调控结果表明, 表面、特别是金属催化剂表面的结构随着反应温度和所处的含不同分子的气氛变化很大. 在催化反应发生的真实条件下, 催化剂表面结构的稳定性首先是受热力学控制, 同时, 随着反应的发生, 还应该是一个动力学过程. 而表面化学为了达到高灵敏度和高分辨率而追求的规整、清洁表面和高真空条件恰恰偏离了催化过程的本质. 为了填补这种基于模型表面的微观高分辨的研究与催化作用的真实结构之间的鸿沟, 科研工作者正致力于发展能在催化反应发生的真实条件下进行高分辨研究的“原位-动态”研究手段和方法, 例如基于同步辐射的光谱和能谱方法、中子谱方法和磁共振方法等[9, 10]. 但是, 现在看来, 这些方法要达到真正从分子水平上理解催化过程所要求的结构上的原子分辨、时间上的纳秒甚至飞秒分辨还有非常大的差距.3 催化中的纳米概念和技术自上世纪末以来, 纳米科学和技术有了长足的进展. 纳米材料的一个重要特性是, 通过改变体系的尺寸如减小到一个特定的范围(如1~100 nm, 根据材料的不同)时, 在不添加任何其他组分的情况下, 体系的电子结构会发生变化. 量子力学已经证明, 大量原子组成的固体材料的价电子为连续的“能带”, 当这类体相材料在某一方向上被缩小, 特别是缩小到纳米尺度时, 电子在该方向的运动就受到空间的束缚和限域, 这种由于限域效应改变电子运动特性将导致体系电子结构特别是价电子结构的改变, 可能会产生量子突变[11, 12]. 这种体系尺寸对电子特性的调变提供了另一条对催化剂的催化特性进行调控的途径. 同时, 大量的催化基础和应用研究也显示, 催化反应事实上就是发生在纳米尺度上, 不论是液相的原子簇(cluster)催化剂, 担载的金属和金属氧化物催化剂, 抑或多孔的分子筛催化剂, 它们体现催化活性的所谓活性中心的尺度就在纳米、甚至亚纳米尺度[13]. 例如, 优良的燃料电池催化剂中贵金属Pt 的粒子平均直径大约2~3 nm, 高分散的Fe 基F-T 合成催化剂中, 活性Fe 物种的最佳尺寸为5~6 nm, 以及具有催化活性的多孔分子筛的孔径一般都小于 1 nm 等等. 这样, 就使研究人员自然而然地将“纳米”与“催化”关联起来, 相信催化过程一定与纳米结构有着本质的联系. 一时间, 大量的催化研究工作者进入到了纳米包信和: 催化基础理论研究发展浅析358研究领域, 相关的文章和报道也大量涌现. 这些研究涉及到了纳米粒子的可控制备、纳米粒子的担载和稳定, 纳米体系的结构和电子特性的表征, 以及纳米催化反应过程等. 总括起来, 这些研究大致可以分为两大类: 一类是纳米粒子的催化基础研究, 主要是发展各类合成方法, 实现具有特定纳结构和电子性质的纳米粒子的可控制备, 借用现代的研究手段, 研究获得的纳米粒子的物理化学性质和对反应分子的吸附、脱附性能. 纵观纳米催化的这类研究, 或多或少地重复了上一世纪表面催化研究的老路, 过分地强调了漂亮的化学(beautiful chemistry), 同样忽略了催化过程的复杂性, 实际反应条件对纳米粒子和相应的模型催化体系的作用, 以及反应体系的热力学平衡和动力学效应等没有给予充分考虑. 纳米催化的另一方面就是针对真实纳米催化体系的研究. 在这类研究中, 纳米粒子的控制制备已经不是很受关注, 而在真实催化反应条件下稳定纳米结构和获得真实条件下纳米催化剂的各类信息变得至关重要. 为了获得纳米催化剂在真实反应条件下的各类参数和特性, 高压扫描隧道显微镜(STM)、环境透射电子显微镜(E-TEM)和基于同步辐射的原位、快速的X-射线吸收谱(XAS), 以及最近的近常压光电子能谱(APHXPS)等相继发展起来, 大量的研究获得了很多出色的研究成果. 与以前的模型表面研究不同, 模型的纳米图4 纳米催化涉及到的材料可控制备、催化应用和基础理论研究等主要内容 粒子与真实催化剂之间有很多可比性, 不存在明显的所谓“物质鸿沟”, 他们的研究结果可以直接拓展到真实催化体系. 因此, 研究人员相信, 只要能发展出在催化反应发生的真实条件下表征催化剂和催化过程的方法和手段, 有效填补这种基础研究和真实过程的所谓“压力鸿沟”, 纳米催化的研究就有可能逼近对催化本质的认识. 因此, 发展表征工具、实现原位、实时和高分辨地表征工作状态下催化剂的研究已经成为了当今催化科学和技术发展面临的又一个新的挑战.4 催化中的限域效应“限域”(c onfinement ), 字面的意思是“被限制在某一区域, 限制的行为或被限制的状态”等, 主要是物理上限制的意思. 这里说的催化中的“限域”, 一个重要特征是通过这种物理上的限制, 主体的特性将发生变化. 比如说, 我们通常知道犯了错误要被“关禁闭”, 这里的“关禁闭”形式上是将对象限定在一个特定(可能是狭小)的空间中, 但本质上是要通过这种物理上的限制, 促使对象反省, 使对象在行为和思想上起变化. 狭义上来说, 催化中的限域效应就是“通过某种物理状态的限制(如纳米状态), 使体系的本征特性(如结构、电子态等)发生变化, 从而改变体系的催化性能”.这方面一个有代表性的例子就是我们近几年来一直致力于研究的有关碳纳米管(CNTs)的限域催化效应[14~17]. 碳纳米管是由石墨烯片以一定的曲率卷曲后, 形成的具有规整的纳米级管腔结构的碳材料. 卷曲过程造成了通常意义上的石墨结构大 键发生畸变, 使电子密度由管内向管外偏移, 从而在管内外形成电势差. 在这种意义上, 碳纳米管的管腔就组成了一种几何上在纳米尺度, 同时又具有独特电子环境的限域体系. 受这种限域系统的作用, 组装在其内部的催化剂活性组分(如纳米粒子)和扩散至腔内的气体分子的特性将会发生变化, 从而导致不同的催化化学. 现在我们已经知道, 这种碳纳米管的限域体系在催化中可能显示三种不同的限域效应[17, 18]. 一是对组装在其管腔中的纳米粒子催化剂: 一方面由于几何空间的限制, 阻碍了其在催化反应过程中的迁移和生长, 另一方面, 由于管腔内的缺电子特性, 使限域其内的纳米粒子的电子特性, 如氧化-还原特性中国科学: 化学 2012年 第42卷 第4期359发生变化. 例如, 组装在4~6 nm 多壁碳纳米管内的Fe 2O 3粒子的还原温度比直接负载在其管外表面要降低近200 ℃. 碳纳米管的另一个限域效应是对反应分子的作用: 由于管腔内外不同的电子环境, 改变了反应分子的吸附能, 造成了反应分子在管内局域浓度的变化. 我们的理论模拟结果显示, 在合成气催化反应中, CO 和H 2在管内富集, 当采用(10,10)双壁管时, 管内外的表观压力相差3~5倍. 这一发现为在温和条件下实现合成气的定向转化提供了理论指南. 碳纳米管的再一个限域效应是对管腔内的催化反应和反应产物. 管腔内独特电子特性调制了特定催化通道的反应能垒, 从而调变了催化反应的选择性. 最近, 中国科学院大连化学物理研究所的李灿研究组报道了采用手性修饰剂辛可尼丁(cinchonidine)修饰限域在碳纳米管管腔中的Pt 纳米粒子, 发现该催化体系在α-酮酯类(ketoesters)的手性氢化反应的活性(TOF)高达1.2×105 h -1, ee 值达到96%, 显著高于碳纳米管管腔外Pt 纳米粒子的催化性能[19]. 进一步的研究发现, 在碳纳米管管腔中, 一些产物分子的扩散特性也发生了非常明显变化. 我们采用超极化129Xe 固体核磁技术研究甲醇分子在碳纳米管中的扩散, 结果表明在碳纳米管管腔中甲醇分子的扩散遵循一种“超扩散”(super diffusion)机理. 当内径为4~6 nm 时, 甲醇分子在碳纳米管内扩散速率是管外的5倍, 是同样管径的硅-铝中孔材料的8~50倍[17]. 这些因素的协同作用, 使具有明显限域性能的碳纳米管材料在未来催化过程的选择调控, 以及未来新催化剂创制的理论和实验研究中呈现出了很大的潜力(图5).这种纳米孔道的限域效应对催化的影响在传统的分子筛催化研究中也能找到很多实证. 现在我们已经知道, 孔径小于1 nm 的分子筛通常会显示出非常明显的限域效应. 实验和理论证明, 这类孔道不但可以作为客体用于稳定组装在其孔道内的纳米粒子, 同时孔道内独特的电子和化学基团的特性也会对其内部催化剂粒子的催化特性起到调变作用, 甚至在这类孔道中有些自身就能显示出非常独特的催化性能. 而当孔径大于1 nm 时, 如中孔硅-铝材料等通常鲜见明显的催化性能, 往往只能作为担载材料用于负载和稳定纳米粒子.最近, 我们在解释氧化亚铁(FeO)纳米岛在金属铂(Pt)表面独特的催化选择氧化性能时引进了一种“界面限域” (interfacial confinement)的概念[20]. 我们的研究发现, 在Pt 表面控制沉积2~5 nm 大小的氧化铁单层小岛, 由于贵金属铂表面与铁原子的相对较强的相互作用, 在特定条件下, 使得Pt 表面上氧化铁物种能稳定保持在低价的氧化亚铁(FeO)状态, 在纳米氧化亚铁岛边缘形成一种配位未饱和的亚铁中心(CUF)(如图6所示). 采用DFT 方法的理论分析表明, 这种结构组合显示出对分子氧的非常高的吸附能力(吸附能为-1.1 eV), 在这种CUF 中心上吸附的分子氧解离形成具有高反应活性的吸附态原子氧物种需要很低的活化能. 在这一高效的催化体系中, 作为衬底的贵金属铂的一个非常重要作用是, 通过与铁原子的强相互作用, 提供了一种本征力, 抗阻了在催化选择氧化条件下原子氧向界面Pt-Fe 键中间的插入. 如果这种插入一旦实现, 表面低价态的FeO 就被深度氧化为配位饱和的高价态, 表面那种高活性的配位未饱和亚铁中心(CUF)将会消失, 催化剂就失去活性. 在这一FeO/Pt 模型体系中, Pt-Fe 界面相互作用导致的这种本征力阻止了活性结构的褪变, 使体系的催化性能得以保持并循环往复. 据此, 我们引出了一种催化限域效应的广义描述, 即催化体系中“一种图5 碳纳米管管腔限域效应包信和: 催化基础理论研究发展浅析360图 6 Pt 表面配位未饱和的亚铁中心(CUF)和氧原子进入Fe-Pt 层间造成了未饱和铁中心的消失本征力(如相互作用力)的存在, 抗阻了体系某种特性发生变化, 或者促使体系变化的特性得以恢复”. 在这种意义上, 催化的限域特性不仅表现为一种现象, 而且抽象成为体系的一种状态和情形.普遍意义上说, 一个优良催化剂必须含有所谓的“活性中心”. 但从能量上来讲, 这种活性中心必然处于高能态, 倾向于通过与反应物的结合, 降低体系能量, 达到稳定态(如形成中间体等). 一旦形成这种稳定的中间体, “活性中心”就失去了继续反应的能力. 因此, 优良催化过程的另一个非常重要的特征就是, 这种稳定态要能被迅速打破, 从而使体系再次回到不稳定的活化态, 放出“活性中心”. 这种从稳定态回到不稳定态的过程需要有一种驱动力. 这种力可以是外部的, 也可以是来自催化剂自身. 大家知道, 这些外部的力可以来自于加热、变压, 也可以来自光、电和磁等外场. 那么催化剂内部是不是也有可能存在这种使活性中心恢复的力? 如存在, 到底有可能是什么? 现在看来, 这种内部的力应该是类似于磁性体系中“矫顽力”, 就是那种抵抗体系变化的本征力. 在作者研究过的催化体系, 特别是低温催化体系中, 这种“矫顽”的能力, 被认为主要来自于体系的限域特性(confinement). 相类似的例子在生物酶催化和化学振荡反应(如Belousov-Zhabotinsky 反应)中很容易找到. 综上所述, 从本质上来说, 催化体系的反应性能不仅仅决定于活性中心的活性, 而且更重要的是决定于体系恢复和再建活性中心的能力, 也就是说促使活性中心恢复, 以及循环往复的那种内禀的本征力.纵观过去一个多世纪催化研究的发展, 人们为打开催化这一“黑箱”进行了大量的实验和理论探索, 从本质上加深了对催化剂和催化过程的认识和理解, 未来的研究将会更加注重发展实时、动态和高分辨的表征工具, 努力获得真实工作状态下催化剂和催化过程的详细信息, 最终实现在原子精度上设计和构筑高效催化剂, 在分子精度上对化学反应过程的进行选择调控.致谢本文的部分内容为作者应邀在第一次中国科学院科学与技术前沿论坛—— “促进可持续发展的催化科学与技术”会议上的报告; 文中的部分实验研究得到国家自然科学基金委重点项目(21033009)的支持; 文章准备过程中得到了中国科学院大连化学物理研究所傅强研究员、李微雪研究员、潘秀莲研究员、杨帆博士、石瑛女士和于良先生, 以及大连理工大学王国豫教授的帮助, 一并致谢!参考文献1 Ertl G, Knözinger H, Weitkamp J, Eds. Handbook of Heterogeneous Catalysis . Weinheim: VCH, 19972 Thomas JM, Thomas WJ. Principles and Practice of Heterogeneous Catalysis . Weinheim: VCH, 19973 Somorjai GA. Introduction to Surface Chemistry and Catalysis . New York: Wiley, 19944 Nilsson A, Pettersson LGM, Nørskov JK. Chemical Bonding at Surfaces and Interfaces . Amsterdam: Elsevier, 20085 Ertl G. Heterogeneous catalysis on the atomic scale. Chem Rec , 2001, 1(1): 33–456 Kitchina JR, Nørskov JK, Barteau MA, Chen JG. Modification of the surface electronic and chemical properties of Pt(111) by subsurface 3dtransition metals. J Chem Phys , 2004, 120: 10240–102467 Xie XW, Li Y, Liu ZQ, Haruta M, Shen WJ. Low-temperature oxidation of CO catalysed by Co 3O 4 nanorods. Nature , 2009, (458): 746–749 8 Somorjai GA, Contreras AM, Montano M, Rioux RM. Clusters, surfaces, and catalysis. Proc Natl Acad Sci , 2006, 103: 10577–10583 9 Salmeron M, Schlogl R. Ambient pressure photoelectron spectroscopy: A new tool for surface science and nanotechnology. Surf Sci Rep ,2008, 63: 169–19910 Tao F, Grass ME, Zhang YW, Butcher DR, Renzas JR, Liu Z, Chung JY, Mun BS, Salmeron M, Somorjai GA. Reaction-driven。

TiO2纳米管内限域纳米Ru及其光催化降解罗丹明B的性能研究余翔;钟颖贤;杨旭;李新军【摘要】采用丙基三甲氧基硅烷(KH570)偶联剂对 TiO2纳米管外表面进行疏水改性,通过浸渍法再经氢气热还原法将 Ru 纳米颗粒原位选择性沉积在 TiO2纳米管内。
采用 TEM、HREM、EDS、HAADF-STEM和紫外可见吸收光谱仪分别对形貌和结构进行表征。
结果表明,内嵌于 TiO2纳米管的 Ru 纳米颗粒粒径约为2~3 nm,TiO2纳米管内负载 w =2% Ru 的光催化性能最好,其光降解罗丹明B 的效率大约是单一 TiO2纳米管的1.8倍。
%The exterior surfaces of the TiO2 nanotube (TNT)were modified by a silane coupling agent to make nano-Ru selectively deposit on the inner wall.The as prepared catalysts were characterized by transmission electron microscope (TEM),high-resolution transmission electron microscopy (HREM), energy dispersive spectrometer (EDS),high-angle annular dark field image (HAADF),scanning trans-mission electron microscopy (STEM)and UV-vis absorption spectra.The results confirm that nano-Ru particles in the range of 2 ~3 nm in diameter are entrapped in the TNTs.TNTs-confined 2% Ru exhibits the best photocatalytic performance,which photocatalytic efficient is 1 .8 times of pure TNTs.【期刊名称】《中山大学学报(自然科学版)》【年(卷),期】2016(055)002【总页数】4页(P85-88)【关键词】水热法;限域催化;TiO2 纳米管;贵金属【作者】余翔;钟颖贤;杨旭;李新军【作者单位】暨南大学生命科学与技术学院化学系,广东广州 510632; 中国科学院广州能源研究所中国科学院可再生能源重点实验室,广东广州 510640;暨南大学生命科学与技术学院化学系,广东广州 510632;中国科学院广州能源研究所中国科学院可再生能源重点实验室,广东广州 510640;中国科学院广州能源研究所中国科学院可再生能源重点实验室,广东广州 510640【正文语种】中文【中图分类】X703环境污染与能源危机是当今世界面临的重要问题,如何解决是摆在人类面前的迫切问题。

纳米碳管在催化剂中的应用研究一、碳纳米管的简介碳纳米管(Carbon nanotubes,CNTs)是一种一维的结构,由碳原子形成纳米尺度的管状物质,在物理、化学、材料科学等领域都具有广泛的应用前景。
碳纳米管单壁的直径通常为1-3 nm,在外径大致相同的情况下,壁厚可以等于单壁厚度,也可以有多壁壁层。
二、纳米碳管在催化剂中的作用催化剂是在化学反应中加快反应速率的物质,它本身并不参与反应过程,而是通过调节反应中的能量变化,实现反应条件的提高,从而促使化学反应的进行。
碳纳米管的结构、性质和表面的化学反应活性使其在催化剂中拥有独特的应用优势。
1. 催化剂支撑材料碳纳米管是一种极其优异的催化剂载体,因其优异的阻塞性能、高比表面积、良好的导电性、高的热稳定性和循环稳定性,使得其可以作为非常理想的催化剂载体来使用。
它可以将催化活性剂稳定地固定在表面上,增加反应过程中的反应基团表面密度,增加反应速率和催化效果。
2. 活性催化剂组分碳纳米管本身也具有催化活性,能够在催化反应中提供表面上的活性位点和催化反应,例如常见的氧化还原反应、还原反应、酯化反应、电荷转移反应等。
在某些反应中,碳纳米管具有比常规催化剂更强的反应选择性,更低的反应温度,更高的催化效率和更快的反应速率。
3. 电催化剂碳纳米管在电化学反应中也具有广泛的应用前景,其能够吸附活性氧和氢气等,从而作为阴、阳极催化剂。
此外,碳纳米管还可以作为超级电容器的核心材料,并且也可以应用在直接甲醇燃料电池中等电化学领域。
三、纳米碳管催化剂研究进展1. 金属催化剂的纳米碳管载体碳纳米管作为金属催化剂的载体具有协同催化作用,为氢化反应、酯化反应、氧化反应等一系列反应提供多种选择。
研究表明,使用纳米碳管作为催化剂载体可以实现对反应活性组分的定向修饰,提高反应性能和催化剂稳定性。
2. 有机功能化纳米碳管催化剂在不同的功能性化物质表面,可以通过非常简单的化学处理方法将这些材料修饰在纳米碳管表面上。

纳米材料在催化反应中的作用原理近年来,随着科学技术的不断发展,纳米材料在各个领域的应用越发广泛。
尤其在催化反应领域,纳米材料的作用备受研究者们的关注。
本文将详细探讨纳米材料在催化反应中的作用原理,旨在揭示其独特的催化效应以及为何纳米材料能够显著改善反应速率和选择性。
一、纳米材料的催化效应纳米材料具有较高的比表面积和高度的晶界活性,这是其展现出卓越催化性能的重要基础。
相较于传统的宏观材料,纳米材料的纳米尺度特征赋予其独特的物理、化学性质,从而展现出以下几个催化效应:1. 尺寸效应纳米材料具有特殊的尺寸效应。
当材料尺寸缩小到纳米级别时,相对表面积的增大使得催化活性位点的数量大幅增加,因而增加了催化反应的活性。
此外,纳米材料较短的传输路径和较低的扩散阻力也有助于提高反应速率。
2. 基底效应纳米材料常常以基底形式存在,即催化活性位点分布在纳米颗粒的表面上。
由于表面活性位点的增多,基底效应能够提高催化反应的速率和效率。
此外,基底效应还可以通过材料的选择性吸附、调节活性位点和提供合适的反应环境等来增强反应选择性。
3. 量子尺寸效应当纳米材料的粒径接近或小于电子波长时,量子效应开始发挥作用。
在纳米材料中,量子效应可以调节电荷分布和电子能级结构,从而改变催化反应的各种动力学和热力学性质。
因此,纳米材料通过量子调控可以实现对反应活性和选择性的精确调控。
二、纳米材料催化反应机制纳米材料在催化反应中的作用原理主要有三种机制,即金属纳米颗粒催化机制、金属氧化物纳米颗粒催化机制和二维纳米材料催化机制。
1. 金属纳米颗粒催化机制金属纳米颗粒催化机制是指金属纳米颗粒作为催化剂参与反应,并通过调整催化活性位点上的电子态以及吸附和解离反应的能力来促进反应。
金属纳米颗粒催化机制被广泛应用于氧化还原反应、还原反应和氧化反应等。
2. 金属氧化物纳米颗粒催化机制金属氧化物纳米颗粒催化机制是指以金属氧化物纳米颗粒作为催化剂进行催化反应。
金属氧化物纳米颗粒具有丰富的氧化还原活性位点,可以参与氧化还原反应、酸碱中和反应等多种反应。

金属纳米粒子的催化作用金属纳米粒子是一种具有特殊性质和潜在应用价值的材料,其催化作用引起了广泛的关注和研究。
本文将从金属纳米粒子的定义、制备方法、催化机理以及应用领域等方面阐述金属纳米粒子的催化作用。
一、金属纳米粒子的定义和制备方法金属纳米粒子是指直径范围在1到100纳米之间的金属粒子。
由于其尺寸效应和表面效应的存在,金属纳米粒子具有与其宏观物质不同的特殊性质,如高比表面积、量子尺寸效应和表面等离子共振等。
制备金属纳米粒子的方法多种多样,常见的方法包括物理法和化学法。
物理法包括溅射法、球磨法和激光蒸发法等,而化学法则是应用广泛的方法,包括还原法、凝胶法和微乳液法等。
二、金属纳米粒子的催化机理金属纳米粒子的催化作用主要源于其特殊的表面性质。
金属纳米粒子具有丰富的表面活性位点和高比表面积,这使得金属纳米粒子能够提供更多的反应活性中心,并提高反应物与催化剂之间的接触效率。
此外,金属纳米粒子还具有量子尺寸效应和电子结构调控效应,这些效应可以调控金属纳米粒子的催化性能。
金属纳米粒子的催化机理可以分为两种类型:金属纳米粒子表面催化和金属纳米粒子内部催化。
对于金属纳米粒子表面催化,反应物吸附在金属纳米粒子表面的活性位点上,通过吸附态的反应物与金属纳米粒子之间的相互作用,发生催化反应。
而金属纳米粒子内部催化是指反应物在金属纳米粒子内部发生反应,通过金属纳米粒子内的空间限制和电子结构调控,加速反应进程。
三、金属纳米粒子的催化应用金属纳米粒子的催化应用十分广泛,包括催化剂、催化剂载体、催化剂修饰剂和催化反应中间体等。
催化剂是金属纳米粒子最主要的应用之一,金属纳米粒子可以作为催化剂用于有机合成、环境治理、能源转化和化学传感等领域。
此外,金属纳米粒子作为催化剂载体,能够提高催化剂的分散性和稳定性,增强催化剂的催化活性和选择性。
金属纳米粒子还可以作为催化剂修饰剂,通过调控金属纳米粒子的形貌、尺寸和表面结构,改善催化剂的性能。

精心整理纳米载体的限域效应对催化性能影响机制的研究进展自上世纪末以来,纳米科学和技术有了长足的进展,其中纳米材料的一个重要特性是,将体系的尺寸减小到一个特定的范围(如1~100nm)时,在不添加任何其他组分的情况下,纳米体系的电子结构会发生变化。
量子力学已经证明,大量原子组成的固图1两种金属催化体系的结构示意图(A)传统的氧化物作为载体的金属催化体系(Oxidesupportedmetalsystem)和(B)过渡金属纳米氧化物倒载型催化体系(oxide-on-metalsystem)如图1所示为传统过渡金属氧化物作为载体的催化体系和过渡金属纳米氧化物倒载型催化体系的结构示意图。
纳米氧化物倒载型催化体系相比传统非均相催化剂,具有更多的TMO/Pt界面(如示意图B中氧化物边缘的黄色虚线所示)。
由于TMO与Pt的表面张力的不同,倒载型催化体系中氧化物(FeO)趋向于在Pt金属表面形成均有双层结构的层状纳米岛结构(由于Fe与Pt具有较强的作用力,双层结构底层与Pt金属结合的为Fe原子,上层为氧原子),而传统催化体系中的Pt金属易于在氧化物颗粒形成较大的颗粒状结构,如下图2所示。
基于上述的界面结构特点,倒载型催化体系具有更多的TMO/Pt界面,并且过渡金属中阳离子(Fe)与贵金属(Pt)间的之间可程造成了石墨结构中大π键的畸变,电子由碳纳米管的凹面向凸面转移,在碳纳米管内外形成一个表观电势差、导致碳纳米管呈现出有别于其他传统碳材料的独特的物理化学特性。
日本富山大学的NoritatsuTsubaki团队在碳纳米管负载铜纳米颗粒催化剂对乙酸甲酯加氢催化过程的研究中,发现了碳纳米管对铜纳米颗粒催化剂的限域效应[3]。
对碳纳米管外壁负载铜纳米颗粒和内壁负载铜纳米颗粒的催化效果进行了对比,由于内壁对于铜纳米颗粒的限域效应非常显着,发现内壁负载的催化剂催化效果明显优于外壁负载的催化剂。
这一限域效应主要表现在:碳纳米管内负载的铜纳米颗粒由于碳纳米管内部的空间限域作用,催化过程中的催化剂颗粒的团聚生长得到有效抑制,从而会防止铜催化剂失活的现象。
碳材料的限域作用我对这碳材料的限域作用啊,还真有自己的一番见解。
我有次去参加一个科技研讨会,会场里坐满了人,一个个都神情专注,有的在奋笔疾书,有的在交头接耳小声议论着。
我看到一个老教授,头发花白如雪,梳得整整齐齐,眼睛在厚厚的镜片后面闪着智慧的光,穿着一件有点皱巴的中山装,正对着台上的报告若有所思地点头。
我就凑过去问:“教授,您能给我讲讲碳材料的限域作用是咋回事吗?”教授看了我一眼,缓缓地说:“这碳材料的限域作用啊,就好比给那些小分子或者离子搭了个特殊的小房子。
你看那碳纳米管,细细长长的,就像一个个精致的小笼子。
一些金属离子进去后,活动范围就被限制住了,就像人被关进了一个狭小的房间,只能在这个限定的空间里活动、反应。
”这时候,旁边一个年轻的研究员也插了话,他穿着一身休闲西装,头发有点乱,眼睛里透着股子兴奋劲儿:“对,还有碳分子筛。
它的孔隙大小不一样,就像不同规格的筛子。
一些气体分子经过的时候,大的分子过不去,小的分子能通过,这就是碳材料的限域作用在起作用,就像交通管制,只允许特定的车辆通行。
我之前做实验,把一些混合气体通过碳分子筛,原本乱糟糟的气体就被分离得井井有条。
”我好奇地问:“那这种限域作用对化学反应有啥影响呢?”老教授推了推眼镜说:“这影响可大了去了。
在限域空间里,反应的活性和选择性都会改变。
就像一群人在大广场和小房间里跳舞,那感觉和效果完全不一样。
在碳材料的限域空间里,有些反应会变得更容易进行,有些则会被抑制。
我曾经研究过一个催化反应,在普通环境下反应效率很低,放到碳纳米管的限域环境里,反应速度一下子就提上去了,就像给一辆慢车换了条快车道。
”正说着呢,一个搞应用开发的工程师也过来了,他皮肤有点黑,看起来很结实,挠挠头说:“那这种碳材料的限域作用在实际应用里怎么利用呢?比如在能源存储方面。
”年轻研究员回答道:“在电池里就可以利用啊。
把一些活性物质放在碳材料的限域结构里,可以提高电池的性能,就像给电池的性能加了个助推器。
碳纳米管在催化剂中的应用近年来,碳纳米管作为一种新兴的纳米材料,逐渐引起了科学家们的关注。
其独特的结构和优异的物化性质,使其具备广泛的应用潜力。
其中,在催化剂领域,碳纳米管也展现出了卓越的性能,被广泛应用于各种催化反应中。
首先,碳纳米管被广泛应用于金属催化剂中。
金属催化剂在有机合成、能源存储等领域具有重要作用。
由于其高比表面积和丰富的表面活性位点,碳纳米管能够提供较优异的载体支撑,增强金属催化剂的稳定性和可再生性。
此外,碳纳米管还能够调控金属纳米粒子的粒径和分散度,进一步提高催化反应的效率和选择性。
其次,碳纳米管在酶催化反应中的应用也备受关注。
酶作为一种高效的生物催化剂,具有特异性、高效性和温和的反应条件等优点。
然而,酶的稳定性和易失活性限制了其在实际应用中的广泛应用。
碳纳米管通过与酶的固定化相结合,可以有效提高酶的稳定性和可重复使用性。
此外,碳纳米管还能够通过调控酶的构象和微环境,影响酶催化活性和选择性。
因此,碳纳米管在生物催化领域具有广阔的应用前景。
此外,碳纳米管还在电化学催化剂中发挥着重要作用。
电化学催化剂广泛应用于燃料电池、电解水制氢等能源转换和储存领域。
碳纳米管作为电子传导性能优异的材料,可作为电化学催化剂的载体或直接参与催化反应。
它能够提供良好的电化学接口,调节电子传递过程,提高催化活性和稳定性。
此外,碳纳米管还具备丰富的官能团,可与不同的活性物种相互作用,进一步改善电化学催化剂的性能。
最后,碳纳米管在环境催化剂中的应用日益受到重视。
碳纳米管能够通过吸附、催化分解等机制,有效去除水中有害物质如重金属离子、有机污染物等。
其高比表面积和孔隙结构可提供较大的吸附容量和反应活性位点,使其在环境治理中具有较大的应用潜力。
综上所述,碳纳米管作为一种有着独特结构和优异性能的纳米材料,在催化剂领域具有广泛的应用前景。
无论是在金属催化剂、酶催化剂、电化学催化剂还是环境催化剂中,碳纳米管都能够发挥出卓越的性能,提高催化反应的效率和选择性。
物 理 化 学 学 报Acta Phys. -Chim. Sin. 2021, 37 (11), 2011027 (1 of 13)Received: November 6, 2020; Revised: November 29, 2020; Accepted: November 30, 2020; Published online: December 7, 2020.*Correspondingauthors.Emails:******************.cn(W.D.);*************.cn(Z.W.).The project was supported by the National Natural Science Foundation of China (22022502, 21776024), the Outstanding Youth Fund Project of Chongqing Natural Science Foundation (cstc2020jcy jjqX0013) and the Program for the Top Young Innovative Talents of Chongqing (02200011130003).国家自然科学基金(22022502, 21776024), 重庆市自然科学基金杰出青年基金(cstc2020jcy jjqX0013)和重庆市青年创新拔尖人才计划(02200011130003)资助项目 © Editorial office of Acta Physico-Chimica Sinica[Review] doi: 10.3866/PKU.WHXB202011027 Regulation of Electrocatalysts Based on Confinement-InducedPropertiesTangfei Zheng, Jinxia Jiang, Jian Wang, Sufang Hu, Wei Ding *, Zidong Wei *Chongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 401331, China.Abstract: The development of highly efficient and low-costelectrocatalysts is important for both hydrogen- and carbon-basedenergy technologies. The electronic structure and coordinationfeatures, particularly the coordination environment and the amountof low-coordination atoms, of the catalyst are key factors thatdetermine their catalytic activity and stability in a particular reaction.The regulation and rational design of catalytic materials at themolecular and atomic levels are crucial to achieving precise chemicalsynthesis at the atomic scale. Recently, significant efforts have beenmade to engineer coordination features and electronic structures byreducing the particle size, tuning the composition of the edges, and exposing specific planes of crystals. Among these representative strategies, the methods based on the confinement effect are most effective for achieving precise chemical synthesis with atomic precision at the molecular and atomic levels. Under molecular or atomic scale confinement, the physicochemical properties are largely altered, and the chemical reactions as well as the catalytic process are completely changed. The unique spatial and dimensional properties of the confinement regulate the molecular structure, atomic arrangement, electron transfer, and other properties of matter in space. It not only adjusts the coordination environments to control the formation mechanism of active centers, but also influences the structural and electronic properties of electrocatalysts. Therefore, the adsorption of catalytic intermediates is altered, and consequently, the catalytic activity and selectivity are changed. In a confined reaction, usually in suitable nano-reactors, the physicochemical properties of reaction products, such as the state of matter, solubility, dielectric constant, and molecular orbital, are finely modulated. Thus, the catalysts produced by confinement significantly differ from those produced in an open system. For example, atomic-layered metals with low coordination can be produced in a two-dimensional confined space. The nitrogen configurations of nitrogen-doped graphene can also be regulated in two-dimensional or three-dimensional confined systems. Herein, the confinement-induced methods, specifically the method used for atomic regulation, are reviewed, such as the control of molecular configuration, the modification of the coordination structure, and the alteration of charge transfer. Applications in the field of fuel cells and material energy conversion are also reviewed. In the next stage, it is important to conduct in-depth investigations of the constructed confinement environment by selecting different substrates for the regulation and rational design of confined catalytic materials. The investigation of the derived properties of the catalyst after release from the confinement is crucial for the development of uncommon catalytic properties.Key Words: Confinement; Electrocatalyst; Electronic structure; Coordinate feature; Molecular configuration. All Rights Reserved.基于限域特性的电催化剂调控郑堂飞,蒋金霞,王健,胡素芳,丁炜*,魏子栋*重庆大学化学化工学院,洁净能源与资源利用化工过程重庆市重点实验室,重庆 401331摘要:开发高效催化剂是促进包括电能源、碳循环等洁净新能源技术发展的关键。
新型纳米材料在催化反应中的应用研究引言催化反应是现代化学工业中的重要过程,它在促进化学反应速率、优化反应条件、提高产率和选择性方面发挥着重要作用。
为了满足可持续发展和环境保护的要求,研究人员一直在寻找更高效、更环保的催化剂。
在这个背景下,新型纳米材料的应用引起了广泛的关注。
本文将着重讨论新型纳米材料在催化反应中的应用研究,包括金属纳米颗粒、纳米孔材料和二维材料等。
一、金属纳米颗粒的应用金属纳米颗粒是一种重要的催化剂,其巨大表面积能够提供更多的反应活性位点,并增强反应物的吸附能力。
金属纳米颗粒广泛应用于氧化反应、还原反应、羰基化反应和烯烃化等多种催化反应中。
例如,铂纳米颗粒在氧化反应中表现出优异的催化活性,可用于催化废水中有毒有机物的加氢脱氧反应。
此外,金属纳米颗粒还可通过形状和尺寸的调控来进一步优化其催化性能。
二、纳米孔材料的应用纳米孔材料是一类具有特殊孔道结构的材料,具有高比表面积和高孔隙容积等特点。
这些特性使纳米孔材料在催化反应中表现出了独特的优势。
例如,具有分子筛结构的纳米孔材料具有尺寸可控的孔道,可用于选择性催化反应。
另外,碳纳米管和金属有机骨架材料等新型纳米孔材料也被广泛应用于氧化、加氢、解聚和还原等催化反应中。
三、二维材料的应用二维材料是具有类似于石墨烯的二维晶体结构的材料,其独特的物理和化学性质使其在催化反应中展现出了巨大的潜力。
二维材料不仅具有高比表面积,而且具有丰富的反应活性位点,使其在催化反应中表现出优异的催化性能。
例如,二维过渡金属硫属化物磷化物材料在氧化、加氢、解聚和还原等催化反应中表现出了卓越的催化活性。
此外,二维材料还可以通过调控其结构和组成进一步优化其催化性能。
结论新型纳米材料在催化反应中的应用研究正逐渐成为化学领域的热点之一。
金属纳米颗粒、纳米孔材料和二维材料等新型纳米材料具有巨大的表面积和丰富的反应活性位点,能够提供更高效、更环保的催化表现。
随着对新型纳米材料催化性能的深入研究,相信将能够进一步发掘新型纳米材料在催化反应中的潜力,并为可持续发展和环境保护等领域的需求提供更好的解决方案。
碳纳米管限域效应
碳纳米管是一种由碳原子构成的纳米结构,具有许多独特的物
理和化学性质。
限域效应是指当材料尺寸减小到纳米尺度时,其性
质会发生显著的变化。
碳纳米管的限域效应是指由于其纳米尺度的
特殊结构,导致其电子输运和热传导等性质受到限制和调控的现象。
首先,从电子输运的角度来看,碳纳米管由于其纳米尺度的特
殊结构,呈现出一维纳米线的特性,使得其电子输运受到限制。
这
种限域效应导致碳纳米管表现出优异的电子输运性能,例如高载流
子迁移率和低电阻率,这使得碳纳米管在纳米电子学领域有着重要
的应用前景。
其次,从热传导的角度来看,碳纳米管的限域效应也对其热传
导性能产生显著影响。
由于碳纳米管的纳米尺度结构限制了热子的
传播,使得其表现出优异的热绝缘性能。
这种特殊的限域效应使得
碳纳米管在纳米材料的热管理和热界面材料方面具有潜在的应用前景。
此外,碳纳米管的限域效应还影响着其化学反应性和力学性能。
由于纳米尺度结构的存在,碳纳米管表现出高比表面积和特殊的表
面化学性质,这使得其在催化和传感等领域具有重要的应用潜力。
同时,限域效应也影响着碳纳米管的力学性能,使得其具有优异的强度和韧性,适用于纳米复合材料等领域。
总的来说,碳纳米管的限域效应在电子输运、热传导、化学反应性和力学性能等方面产生重要影响,这使得碳纳米管在纳米科技领域具有广泛的应用前景。
对于科学研究和工程应用而言,深入理解和控制碳纳米管的限域效应具有重要意义。
附件2论文中英文摘要格式作者姓名:陈为论文题目:碳纳米管限域的金属纳米粒子的催化行为作者简介:陈为,男,1977年7月出生,2003年9月师从于中国科学院大连化学物理研究所包信和研究员,于2008年3月获博士学位。
中文摘要随着石油价格的高涨及其资源的日益枯竭,迫使人们寻找新的清洁、可持续的能源替代产品。
以煤和天然气为资源经合成气催化转化成液体燃料是一种非常有应用前景的过程,对于保障我国能源安全及解决环境污染问题等都具有重大的经济和现实意义,发展高效催化合成气转化的催化剂显得越来越紧迫和重要。
碳纳米管自1991年被Iijima发现以来,因其独特的结构和性能引起了人们极为广泛的关注,尤其是碳纳米管的纳米级管道为纳米粒子提供了准一维的限域环境。
本论文研究了碳管的限域环境对Fe/Fe2O3粒子的氧化还原性能的调变作用,以及这种限域效应对F-T合成反应性能的影响,取得了如下结果:1. 发展了高效的碳纳米管填充方法—湿法毛细诱导填充法尽管各种填充方法日趋成熟,然而现有的很多碳纳米管填充的复合体系并不适合于催化应用,如原位填充的金属及其化合物完全被密封在碳纳米管管腔中;熔融填充的金属纳米线或纳米棒严实地充满整个碳纳米管内腔,大部分金属并不能与外界接触;Green等开创的湿化学填充法,尽管能得到颗粒状填充的过渡金属,但是这个方法对金属盐的消耗量较大,不适用于填充贵金属,并且无法准确定量。
这些填充方法的填充效率高低不一,并且其填充复合物的产量还不能够达到一般催化剂量的要求。
因此,发展一种适用于催化应用的普适性强的、高效的填充碳纳米管的方法,是实现碳纳米管的“管中催化”亟需解决的首要问题。
相对于其它填充碳纳米管方法,湿化学填充法简单,可得到颗粒状填充的过渡金属粒子。
我们针对湿化学填充碳纳米管的方法存在填充效率不高、不易准确定量的缺点,结合碳管本身的结构特点进行了改进,发展了湿法毛细诱导填充法。
主要步骤是:首先将碳纳米管端口打开,同时进行表面亲水性处理,使得碳管能够被溶液完全浸润;然后,利用强超声振荡下的空化作用,使碳管内的残余物能够扩散出来,从而含金属离子的溶液能够在毛细力作用下进入碳管管腔;最后,控制溶液的蒸发速率,金属离子在浓度差的驱动下,尽可能进入到碳管管腔中,之后加热使金属前驱物发生分解,得到金属氧化物填充的碳纳米管复合物。
这种方法适用于多种金属催化剂在碳纳米管中的填充,具有普适性。
TEM显示80%以上的Fe2O3纳米粒子都进入了碳管内腔。
这为充分利用碳纳米管所独有的准一维纳米环境及其限域效应,提高碳纳米管在催化领域中的应用水平,提供了技术支持。
2.发现了碳纳米管对限域的Fe/Fe2O3氧化还原性能的调控效应许多研究发现碳纳米管内填充的物质在物理、化学等诸多性质上发生了改变。
然而,涉及到多相催化活性组分主体的金属(或氧化物)在碳管内的氧化还原性质的研究却鲜见报道。
在多相催化领域,催化剂活性组分的氧化还原性能及其化学稳定性对催化剂的活性、选择性以及在反应过程中的稳定性有着重要的影响。
碳纳米管可看作是石墨烯片以一定的曲率卷曲后,形成具有规整的纳米级管腔结构的碳材料。
卷曲过程造成了通常意义上的石墨结构中大π键发生畸变, 使得π电子云由管内向管外偏移, 从而在管内外形成一个表观电势差,管内的电子云密度相对小于管外。
这样若将金属纳米粒子分散于碳管管腔内,其所处的电子环境将不同于碳管外壁,若金属粒子与碳管壁之间存在相互作用,就有可能修饰金属粒子的电子结构,并影响其氧化还原性能。
我们首次将长波长激光Raman光谱用于表征碳纳米管管腔内的金属氧化物纳米粒子。
这为研究限域于碳纳米管管腔中的纳米氧化物复合体系提供了一种有力的技术手段。
Raman光谱显示,限域的Fe2O3中Fe-O振动峰随碳管内径的减小而发生蓝移,表明了碳管管壁与Fe2O3粒子之间越来越强的相互作用。
采用多种表征手段,研究了分别负载于管内外的Fe和Fe2O3的氧化还原性质,发现碳纳米管的限域环境对Fe/Fe2O3纳米粒子氧化还原性能具有调变作用。
通过原位加热HRTEM、原位XRD、原位拉曼和程序升温升温实验发现,内径为4-8 nm的碳纳米管内的Fe2O3粒子在600°C时即可被碳还原生成金属Fe,而碳管外粒子的还原温度为800°C。
且碳管内的Fe2O3纳米粒子的还原温度随着碳管内径的减小而逐渐降低。
原位XRD实验还发现,当还原剂为氢气,一氧化碳时,碳纳米管内的Fe2O3纳米粒子同样比管外粒子更容易还原。
我们采用原位拉曼和微天平对金属Fe在碳纳米管内外、活性碳、SiO2上以及SBA-15孔道中的氧化过程进行了对比研究,发现金属Fe发生氧化反应的顺序(由难到易)为:inside CNT > outside CNT ≈ on XC-72 > inside SBA ≈ on SiO2,即限域于碳管管道中的金属Fe的氧化反应滞后发生。
通过DFT理论计算,证实了碳纳米管内外的电势差可以造成碳管(i.d. 3 nm)内外的Fe2O3粒子的还原温度相差最大可达200°C,这与实验结果基本吻合。
3.证实了碳纳米管限域的Fe催化剂对F-T合成反应性能具有促进作用将碳纳米管限域的纳米金属Fe催化剂用于F-T合成反应,考察了碳管的限域作用对催化性能的影响。
通过对负载于碳纳米管内外的两种金属Fe催化剂Fe-in-CNT和Fe-out-CNT作对比研究,发现Fe-in-CNT的活性和C5+烃类产物时空收率均高于Fe-out-CNT。
我们在原位XRD的反应腔中,模拟真实F-T合成反应过程,发现碳管的限域作用提高了管内粒子的可还原性;切换到合成气气氛下,金属铁以及其它铁物种与合成气作用生成了活性组分碳化铁,碳管内的碳化铁物种的比例高于管外粒子;升高反应压力后,碳化铁物种的比例也几乎不变。
整个反应过程中碳管内的活性组分碳化铁始终保持着较高的比例,显示出较高的反应活性。
同时碳管管腔的空间限制作用也有利于碳链的增长,使得长链烃产物的选择性较高。
我们还将Rh-Mn纳米粒子组装到碳管管道内,用作合成气转化制C2含氧化合物反应过程的催化剂。
发现碳纳米管管腔对金属粒子具有空间限制效应,可限制粒子的进一步长大,从而保持反应活性稳定。
这对于结构敏感性反应,控制催化剂粒子大小提供了一种新思路。
同时为金属催化剂,尤其是贵金属经常面临的烧结问题提供了解决方案。
原位Raman实验表明,碳管内还原的催化剂吸附CO后,含有较多的倾斜式吸附的CO物种,有利于CO分子的解离和C2含氧化合物的生成,因此管内催化剂的C2含氧化合物的生成活性高于管外催化剂。
这类复合催化剂上所表现出的独特催化性能为碳管和金属纳米粒子体系的“协同限域效应”所致。
这给我们一个启示,可以利用碳纳米管的限域效应改善或者提高涉氢的多相催化反应的催化性能,因为这些催化反应中低价态的金属是反应的活性物种。
关键词:碳纳米管,催化,氧化铁,自还原,氧化还原,限域效应,碳化铁,F-T合成Catalytic behaviors of metal nanoparticles encapsulated inside carbonnanotubesChen WeiABSTRACTWith soaring oil prices and dwindling resources, it is urgent to screen out clean and sustainable fuel alternatives. The synthesis of liquid fuel based on Fischer-Tropsch (F-T) synthesis through coalor natural gas would be of significance to resolve the problems of the national energy-supply security and environment protection. On the other hand, carbon nanotubes (CNTs) have been used as supports widely in catalysis due to their unique structure and properties since they were discovered by Iijima in 1991. In particular, the seamless nanochannels formed from graphene layers may provide a unique one-dimensional confinement environment for nanocatalysts and catalytic reactions. In this thesis, the influence of the confinement within CNTs on the structure and redox properties of encapsulated Fe/Fe2O3 nanoparticles, and further on catalytic performance has been investigated. In summary, we obtained the following results:1. We established a highly efficient method to introduce and disperse transition metal oxide particles into the CNT channels.Although extensive studies have been undertaken to introduce transition metal oxides into the CNT channels, most of as-formed composites are not applicable in catalysis. For example, the encapsulated metals through the in situ filling method are often isolated completely inside the nanotubes, which will be inaccessible to reactants in catalytic reactions. Melted salt method produces nanowires or nanorods generally that block the CNT channels. Although nanoparticles can be obtained inside CNTs through the wet chemistry method proposed by Green et al., it is inconvenient for controlling the metal loadings. Therefore, it is urgent to develop a universal and efficient method to fill CNTs.Based on the wet chemistry method, we refluxed CNTs in concentrated nitric acid to obtain open-end CNTs decorated by surface oxygen groups, then introduced the solution containing metal precursors into the CNT channels utilizing the capillary forces of CNTs aided by ultrasonication and stirring. Subsequent drying and heat treatment resulted in CNT-encapsulated transition metal oxide nanoparticles.TEM showed that over 80% nanoparticles were located inside the CNT channels. These processes are applicable to many other metals and metal oxides and thus provide the basis for further experimental studies of the CNT confinement in catalysis.2. We reported that the encapsulation within CNT nanochannels had a significant influence on the redox properties of iron and iron oxide.Many studies have shown the unusual physiochemical properties of encapsulated transition metal nanoparticles inside CNTs, however, there is a lack of knowledge the redox behaviors andchemical stability of the encapsulated transition metals, which are essential for the applications in catalysis.Deviation from planarity causes rehybridization to become intermediate between sp2 and sp3 in the graphene layers, and as a result π-electron density is shifted from the concave inner to the convex outer surface of CNTs. Thus the interior surface becomes electron-deficient while the outer surface becomes electron-enriched. When a transition metal is placed in the CNT interior, it should interact with the grapheme surface in a different way from that located on the CNT exterior.The interaction of the confined iron oxide with CNTs was characterized by Raman spectroscopy. Fe-O band was blue-shifted when the Fe2O3 particles were moved from the exterior to the interior of CNTs. It further blue-shifted when the inner diameter became smaller. It implied the presence of an interaction of the Fe2O3 particles with the inner CNT surface. Furthermore, this interaction becomes stronger within smaller CNTs.Characterization by in situ HRTEM, in situ XRD, in situ Raman spectroscopy and temperature-programmed reaction revealed that Fe2O3nanoparticles confined inside CNTs (inner diameter 4–8 nm) were reduced by carbon to metallic iron at 600 °C while the reduction temperature of the outside iron oxide was 800 °C. Interestingly, the reduction temperature of the confined Fe2O3 decreased with the smaller inner diameter of CNTs. It showed that CNT-confined Fe2O3 were also easier reduced in H2 and CO atmosphere compared to outer Fe2O3.Oxidation of CNT-confined metallic Fe particles was found to be retarded in comparison to that of outer Fe particles, as shown by in situ XRD and gravimetric measurements with an online microbalance. The oxidation temperature of Fe particles decreased in the following order: inside CNT > outside CNT ≈ on XC-72 > inside SBA ≈ on SiO2.DFT theoretical studies indicated that the potential energy inside nanotubes was lower than that outside CNTs, which could lead to a lower reduction temperature. It agreed well with experimental results.3. We found the effect of the confinement within CNTs could improve the activity of F-T synthesis reaction.The Fe catalyst confined inside the CNTs shows better catalytic performance compared to that dispersed on the outer walls of CNTs. The CO conversion and yield of C5+ hydrocarbons over the encapsulated iron catalyst were higher than that on the outer iron catalyst. This was confirmed by in situ XRD characterization when iron was exposed to syngas under FTS conditions. TheCNT-confined iron catalyst exhibited an obviously higher iron carbide/iron oxide ratio in comparison to the outside catalyst. This was due to the interaction of metallic iron with dissociated CO. The improved reducibility favored formation of more iron carbides, which is responsible for the higher activity over the CNT-confined iron catalyst. The CNT channels are also beneficial for the chain growth of the intermediates, leading to a higher selectivity to longer chain hydrocarbons C5+.A bicomponent RhMn catalyst was introduced into the CNT channels for synthesis C2 oxygenates from syngas. A remarkable enhancement of the catalytic activity was observed compared to the catalyst with RhMn dispersed on the CNT exterior surfaces. Characterization of the catalysts with TEM showed that the channels of CNTs had space restriction on RhMn catalysts, which effectively prevented the sintering of metal particles during the reaction. Raman characterization of the CO-adsorbed catalysts indicated that the activation of CO was likely modified inside CNTs. CO tend to adsorb in a titled form on RhMn catalyst confined in CNTs due to the electron-deficient interior surface of CNTs, which facilitates the dissociation of CO and the formation of C2 oxygenates.The enhanced activities on CNT-confined catalysts are due to the synergetic confinement inside CNTs. This provides a novel approach to tune the catalytic behaviors of metal catalysts for the involving hydrogen reactions, which are sensitive to the electronic state of the active components.Key words: Carbon nanotubes, catalysis, iron oxide, autoreduction, redox, confinement effect, iron carbide, F-T synthesis。