氯吡格雷的概况
- 格式:doc
- 大小:55.16 KB
- 文档页数:9

氯吡格雷用于冠心病治疗的效果以及不良反应氯吡格雷是一种抗血小板药物,广泛应用于冠心病患者的治疗中。
它可以帮助预防心脏血管疾病的进展和发展,从而减少心脏事件的风险。
但氯吡格雷也可能会引起一些不良反应。
本文将重点介绍氯吡格雷在冠心病治疗中的效果以及可能出现的不良反应。
效果临床研究表明,对于冠心病患者,氯吡格雷可以显著减少心肌梗死、中风和其他心脏事件的发生。
尤其是对于有急性冠脉综合征的患者来说,氯吡格雷更是必不可少的治疗药物之一。
大规模的临床试验已经证实,氯吡格雷能够显著降低急性冠脉综合征患者的心脏事件发生率,同时不增加出血的风险。
除了急性冠脉综合征外,对于稳定型冠心病患者来说,氯吡格雷也具有一定的治疗作用。
一些临床研究显示,长期服用氯吡格雷可以减少稳定型冠心病患者的心肌梗死和心脏事件的发生率,延缓疾病的进展。
不良反应尽管氯吡格雷在预防冠心病事件方面表现出色,但也不能忽视其可能带来的不良反应。
在临床上,常见的氯吡格雷不良反应主要包括出血、胃肠道不适和过敏反应等。
出血是氯吡格雷的最主要不良反应之一。
由于氯吡格雷能够影响血小板的凝血功能,从而增加出血的风险。
一些临床研究显示,长期使用氯吡格雷会导致消化道出血、脑出血、皮肤瘀斑等出血事件的发生率增加。
尤其是对于老年患者和合并其他出血风险因素的患者来说,需要特别小心使用氯吡格雷。
胃肠道不适也是氯吡格雷常见的不良反应之一。
包括恶心、呕吐、腹泻、消化不良等症状。
这些胃肠道不适的出现可能会影响患者对氯吡格雷的依从性,从而降低药物的疗效。
过敏反应是一些患者在使用氯吡格雷时可能出现的不良反应。
轻微的过敏反应可能表现为皮疹、瘙痒、荨麻疹等症状,严重的过敏反应则可能引起呼吸困难、心跳加快、血压下降等严重表现。
对于存在对氯吡格雷过敏史的患者来说,禁忌使用氯吡格雷。
结语氯吡格雷作为冠心病治疗的重要药物,其预防心脏事件的效果显著,受到临床医生和患者的广泛认可。
但不良反应也是需要引起足够重视的。

氯吡格雷的描述氯吡格雷,这可真是个神奇的小药片呀!你可别小瞧了它,它在咱们的健康领域里那可是有着相当重要的地位呢。
咱就说,这身体啊就像一部精密的机器,有时候血管里会出现一些“小状况”,就好比道路上出现了一些障碍物。
而氯吡格雷呢,就像是个勤劳的“清道夫”,能帮着把这些障碍给清理掉,让血液能顺畅地流动。
你想想看,要是血管里堵了东西,那多吓人呀!这就好像家里的水管被堵住了,水都流不出来了,那家里还不得乱套呀!氯吡格雷就能在这种关键时刻发挥大作用呢。
它能抑制血小板的聚集,就像是给那些调皮捣蛋的血小板套上了一个“紧箍咒”,让它们别乱跑乱聚,乖乖地保持秩序。
这多重要啊,不然血小板们瞎捣乱,那后果可不堪设想。
很多人可能会问,那吃这个药有没有啥要注意的呀?嘿,那当然有啦!就像你去参加一个重要活动,总得知道些规矩吧。
首先呢,得听医生的话,医生让怎么吃就怎么吃,可别自己瞎捣鼓。
医生那可是专业的,他们知道怎么给你安排最合适。
还有啊,吃药期间也得注意自己的身体反应。
要是感觉有啥不对劲的,比如身上起疹子啦,或者觉得头晕乎乎的,那可得赶紧跟医生说呀!这就跟你走路似的,要是路上有个坑,你得赶紧绕过去或者找人来填上,可不能装作没看见就踩进去了呀。
而且呀,吃氯吡格雷的时候可别乱吃药。
有些药跟它一起吃可能就会出问题,就像两个合不来的人凑到一块,肯定会闹别扭呀。
所以呀,一定要把自己吃的药都跟医生交代清楚,让医生来判断能不能一起吃。
咱再说说这药的效果。
那可真是立竿见影呀!很多人吃了之后,身体的状况明显就改善了。
就好像原本昏暗的房间一下子打开了窗户,阳光照进来了,那感觉多好呀!总之呢,氯吡格雷是个好东西,但咱也得正确地使用它。
就像一把好刀,得在会用的人手里才能发挥出最大的作用。
咱可不能浪费了这么好的药,得让它为咱的健康好好保驾护航。
所以呀,大家一定要重视起来,和医生好好配合,让氯吡格雷成为我们健康的小卫士!原创不易,请尊重原创,谢谢!。

氯吡格雷代谢基因氯吡格雷是一种抗血小板药物,常用于预防心脑血管疾病,例如心脏病和中风。
然而,每个人对药物的反应不同,其中一部分原因可能是由于个体的代谢基因差异。
首先,氯吡格雷主要通过肝脏酶系统代谢。
酶是一种催化化学反应的蛋白质,它们帮助将药物分解为代谢产物,以便能够在体内被排出。
对于氯吡格雷来说,其中一个关键的代谢酶是CYP2C19。
这个酶的活性有可能受到个体基因的表达水平的影响。
研究表明,CYP2C19基因有多个变异型。
其中,CYP2C19*1型被认为是正常活性的基因,而CYP2C19*2和CYP2C19*3则是常见的变异型。
这些变异型导致CYP2C19酶的活性降低,从而使得氯吡格雷的代谢速率下降。
因此,携带CYP2C19*2或CYP2C19*3的个体可能需要更低的氯吡格雷剂量来达到相同的药效。
此外,还有其他一些CYP2C19变异型,如CYP2C19*4、CYP2C19*5和CYP2C19*17等。
这些变异型对CYP2C19酶的活性也产生了不同程度的影响。
其中,CYP2C19*17型的表达与酶的活性升高相关,这可能导致氯吡格雷的代谢速度加快,从而需要更高的剂量才能达到预期的治疗效果。
了解个体的氯吡格雷代谢基因类型可以为个体化药物治疗提供有益的信息。
一些研究发现,携带CYP2C19变异型的个体在接受氯吡格雷治疗时可能更容易出现药物耐受性和治疗失败。
因此,对于这些患者,可能需要调整药物剂量或尝试其他的抗血小板药物。
总结而言,氯吡格雷代谢基因的变异可能对个体对药物的反应产生影响。
了解个体的CYP2C19基因型可以为氯吡格雷的药物治疗提供指导。
未来,个体化药物治疗的发展将依赖于对代谢基因的深入了解,从而为患者提供更有效的个性化治疗方案。

预防脑卒中的氯吡格雷,什么时候需要停药?医师讲出实情提起来氯吡格雷,想必大家并不陌生。
与阿司匹林不同的是,氯吡格雷通过拮抗P2Y12受体,抑制血小板的聚集,进而预防血栓的形成。
临床上常用于预防心肌梗死,缺血性脑卒中等血栓事件的发生。
对于存在阿司匹林抵抗、阿司匹林禁忌症以及服用阿司匹林后出现不耐受的情况时,可以考虑换用氯吡格雷来进行替代。
众所周知,人体内的血小板每天都是在不停更新的。
因此,不论是阿司匹林还是氯吡格雷都需要长期服用,以此降低脑卒中的发生风险。
然而,是药三分毒,氯吡格雷也不例外,服药过程中如果出现以下两种情况时,需要及时停用。
一、手术前
对于近期需要做手术范围比较大的外科手术时,临床上会建议患者停用氯吡格雷5-7天。
这主要是由于脑卒中患者每天服用75mg的氯吡格雷后,对血小板的抑制作用在服药后3-7天达到稳态。
而治疗终止后血小板聚集和出血时间约在5天的时间内逐渐回到基线水平。
二、出血
氯吡格雷会抑制血小板衍生的生长因子和血小板血管内皮生长因子的释放,导致新生血管的生成过程受阻,进而使得胃肠道黏膜的保护和修复作用丧失,增加胃肠道出血的风险。
因此,如果服用氯吡格雷过程中出现大便发黑,暗红色血便、呕血等情况,应及时停用该药物。
此时,建议可以服用雷贝拉唑或者泮托拉唑来保护胃粘膜,减轻胃肠道损伤。
药物的副作用是药物在治疗疾病过程中不可避免产生的,这就提醒我们大家,在治疗疾病的同时也要关注药物不良反应的发生,以免对机体产生损害。

每日医药——氯吡格雷
【其他名称】波立维。
【性状】常用其二硫酸盐,为白色粉末,不溶于水,但于ph1的水中易溶。
易溶于甲醇。
【药理及应用】是血小板聚集抑制剂,选择性的抑制ADP与血小板受体的结合及抑制ADP介导的糖蛋白GPⅡb/Ⅲa复合物的活化,而抑制血小板聚集。
也可抑制非ADP引起的血小板聚集。
对血小板ADP受体的作用是不可逆的。
口服吸收迅速,血浆中蛋白结合率为98%,在肝脏代谢,主要代谢产物无抗血小板聚集作用。
用于预防和治疗因血小板高聚集引起的心、脑及其他动脉循环障碍疾病,如近期发作的脑卒中、心肌梗死和确诊的外周动脉疾病。
【用法】每日一次,每次75mg。
【注意】(1)常见的不良反应为消化道出血、中性粒细胞减少、腹痛、食欲减退、胃炎、便秘、皮疹等。
偶见血小板减少性紫癜。
(2)对本品过敏者禁用。
(3)溃疡病患者及颅内出血患者禁用。
(4)肝、肾功能损害者慎用。
【制剂】片剂:每片25mg;75mg。


氯吡格雷(Clopidogrel)使用说明书氯吡格雷(Clopidogrel)使用说明书氯吡格雷(Clopidogrel)是一种用于预防血栓形成的药物。
本使用说明书将为您提供关于氯吡格雷的详细信息,包括药物的作用、用法、剂量和潜在的副作用。
在使用本药物之前,请仔细阅读本说明书,并按照医生或药师的指导使用。
一、药物作用氯吡格雷属于一类称为ADP受体拮抗剂的药物。
它通过抑制血小板聚集和血栓形成来预防心血管事件的发生,如心肌梗死和中风。
氯吡格雷通过抑制ADP受体,阻断ADP对血小板的刺激作用,从而减少血小板的聚集和血栓的形成。
二、适应症氯吡格雷用于以下情况的预防:1.缺血性心脏病:对于患有稳定性冠心病或急性冠状动脉综合征的患者,氯吡格雷可用于预防心肌梗死、中风和血栓形成。
2.急性冠状动脉综合征:在心脏血管介入手术(如冠状动脉支架植入术)前后,氯吡格雷可以降低血栓形成和再狭窄的风险。
3.出血性中风:对于已经发生过非出血性中风的患者,氯吡格雷有助于预防再次中风的发生。
3.用法和剂量请遵循医生或药师的建议,并按照正确的剂量和用法使用氯吡格雷。
通常情况下,成年人每天口服一次氯吡格雷75毫克,建议在饭后服用,以减少胃肠道不良反应的发生。
务必不要超过医生建议的剂量,也不要停止使用药物,除非经过医生的指导。
如果错过一次剂量,请尽快补充剂量,但不要一次服用双倍剂量。
4.注意事项在服用氯吡格雷期间,请注意以下事项:- 如果您需要进行手术或牙科手术,请提前告诉医生或牙医,因为氯吡格雷可能会影响止血能力。
- 如果您出现任何出血或瘀伤的症状,如鼻出血、牙龈出血或皮肤瘀伤,请立即告知医生。
- 如果您正在服用其他药物,请告知医生或药师,因为某些药物可能与氯吡格雷发生相互作用。
- 如果您怀孕、哺乳或计划怀孕,请在使用氯吡格雷之前告知医生。
- 氯吡格雷可能引起一些副作用,如胃肠道不良反应、头痛和乏力。
如果您遇到任何副作用,应及时告知医生。
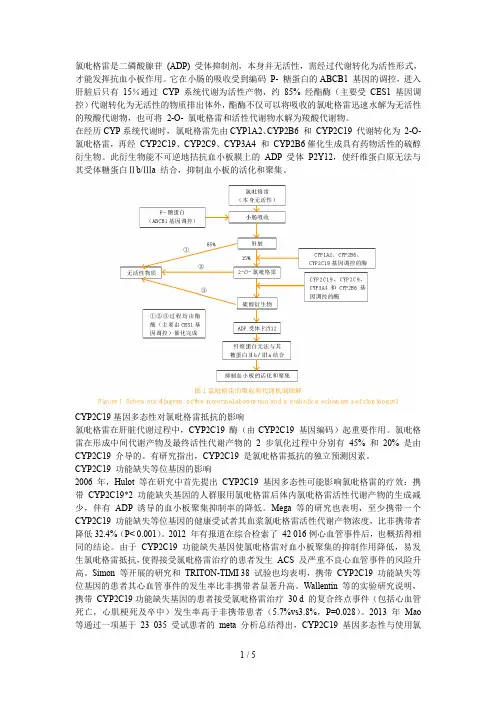
氯吡格雷是二磷酸腺苷(ADP) 受体抑制剂,本身并无活性,需经过代谢转化为活性形式,才能发挥抗血小板作用。
它在小肠的吸收受到编码P- 糖蛋白的ABCB1 基因的调控,进入肝脏后只有15%通过CYP系统代谢为活性产物,约85% 经酯酶(主要受CES1基因调控)代谢转化为无活性的物质排出体外,酯酶不仅可以将吸收的氯吡格雷迅速水解为无活性的羧酸代谢物,也可将2-O- 氯吡格雷和活性代谢物水解为羧酸代谢物。
在经历CYP系统代谢时,氯吡格雷先由CYP1A2、CYP2B6 和CYP2C19 代谢转化为2-O- 氯吡格雷,再经CYP2C19、CYP2C9、CYP3A4 和CYP2B6催化生成具有药物活性的硫醇衍生物。
此衍生物能不可逆地拮抗血小板膜上的ADP 受体P2Y12,使纤维蛋白原无法与其受体糖蛋白Ⅱb/Ⅲa 结合,抑制血小板的活化和聚集。
CYP2C19基因多态性对氯吡格雷抵抗的影响氯吡格雷在肝脏代谢过程中,CYP2C19 酶(由CYP2C19 基因编码)起重要作用。
氯吡格雷在形成中间代谢产物及最终活性代谢产物的 2 步氧化过程中分别有45% 和20% 是由CYP2C19 介导的。
有研究指出,CYP2C19 是氯吡格雷抵抗的独立预测因素。
CYP2C19 功能缺失等位基因的影响2006 年,Hulot 等在研究中首先提出CYP2C19基因多态性可能影响氯吡格雷的疗效:携带CYP2C19*2 功能缺失基因的人群服用氯吡格雷后体内氯吡格雷活性代谢产物的生成减少,伴有ADP 诱导的血小板聚集抑制率的降低。
Mega 等的研究也表明,至少携带一个CYP2C19 功能缺失等位基因的健康受试者其血浆氯吡格雷活性代谢产物浓度,比非携带者降低32.4%(P< 0.001)。
2012 年有报道在综合检索了42 016例心血管事件后,也概括得相同的结论。
由于CYP2C19 功能缺失基因使氯吡格雷对血小板聚集的抑制作用降低,易发生氯吡格雷抵抗,使得接受氯吡格雷治疗的患者发生ACS 及严重不良心血管事件的风险升高。

氯吡格雷用于冠心病治疗的效果以及不良反应1. 引言1.1 氯吡格雷是一种常用的抗血小板药物氯吡格雷是一种常用的抗血小板药物,也被称为氯吡格雷。
它是一种ADP受体拮抗剂,主要通过抑制ADP所致的血小板聚集,从而减少血栓形成的可能性,是一种有效的抗血小板药物。
氯吡格雷在临床上被广泛应用于冠心病的治疗中,特别是在急性冠脉综合征的治疗中具有重要的地位。
它可用于预防血栓形成,减少心肌梗死和缺血性中风的发生,同时也可用于治疗稳定性冠心病患者。
由于其良好的疗效和安全性,氯吡格雷被认为是冠心病治疗中不可或缺的一部分。
医生在治疗冠心病时常常会选择氯吡格雷作为血小板抑制剂的首选药物之一。
氯吡格雷的广泛应用为冠心病患者提供了良好的治疗选择,有效改善了患者的生活质量和预后。
1.2 氯吡格雷在冠心病治疗中的作用氯吡格雷是一种常用的抗血小板药物,被广泛用于冠心病治疗。
冠心病是一种心血管疾病,是由冠状动脉狭窄或阻塞导致心肌缺血、缺氧的病变。
氯吡格雷通过抑制血小板聚集和凝血酶原的活化,阻断血栓形成,可以有效预防冠心病发作。
氯吡格雷还可以降低心衰、心律失常等心血管并发症的发生风险,提高患者的生存率。
氯吡格雷在冠心病治疗中起着关键作用,特别是在急性冠状动脉综合征、心梗、支架植入等情况下,氯吡格雷的使用可以显著减少心血管事件的发生率,改善患者的预后。
临床研究表明,与单独使用阿司匹林相比,联合使用氯吡格雷可以进一步降低心血管事件的风险,提高患者的生存率和生活质量。
氯吡格雷在冠心病治疗中具有重要的作用,是不可或缺的药物之一。
在临床实践中,医生应根据患者的具体情况,合理使用氯吡格雷,以达到最佳的治疗效果。
2. 正文2.1 氯吡格雷对冠心病的治疗效果氯吡格雷是一种常用的抗血小板药物,被广泛用于冠心病的治疗。
其主要作用是通过抑制血小板凝集,阻止血栓的形成,从而预防心血管事件的发生。
研究表明,氯吡格雷在冠心病患者中能够显著降低心血管事件的风险,如心肌梗死、脑卒中等。

基本药物处方一氯毗格雷【制剂规格】75mg∕片【适应症】可用于防治心肌梗死,缺血性脑血栓,闭塞性脉管炎和动脉粥样硬化及血栓栓塞引起的并发症。
应用于有过近期发生的中风、心肌梗死或确诊外周动脉疾病的患者,治疗后可减少动脉粥样硬化事件的发生(心肌梗死、中风和血管性死亡)。
【不良反应】常见的不良反应有皮疹(4%)、腹泻(5%)、腹痛(6%)、消化不良(5%)、颅内出血(0.4%)、消化道出血(2%),严重粒细胞减少(0∙04%)°与阿司匹林相似。
主要是大型临床试验(CAPRIE)中评价的结果。
该研究中氯口比格雷的总体耐受性与ASA相当,与年龄、种族和性别无关。
出血性疾病:胃肠道出血、紫瘢、淤血、血肿、鼻蚓、血尿、眼出血(主要是结膜出血)和颅内出血。
氯毗格雷治疗病人的严重出血发生率为1.4%。
血液系统:包括严重中性粒细胞减少,再生障碍性贫血和严重血小板减少,均比较罕见。
【用法用量】波立维的推荐剂量为每天75mg,心血管疾病症状不是很明显,可2-3天服一次,就餐结束前与食物同服可减少对胃的刺激程度。
【药物相互作用】华法林:见注意事项。
肝素:在健康志愿者的研究中,氯毗格雷不改变肝素在凝血上的作用,不必要改变肝素的剂量。
同时服用肝素不影响氯毗格雷诱导的对血小板聚集的抑制效果。
由于同时服用的安全性没有确定,因此使用应谨慎。
血栓溶解剂:近期发作的心肌梗死的病人,同时服用氯毗格雷,K-PA和肝素,评价其安全性。
临床出血的发生率与rt-PA和肝素同阿司匹林同时服用的发生率相似。
由于氯此格雷与其他血栓溶解剂同时服用的安全性没有确立,因此使用时应谨慎。
([注意事项])非留体解热镇痛药(NSALDS)健康志愿者同时服用蔡普生和氯毗格雷与潜在的胃肠道出血有关,由于缺少氯吐格雷与其他非将体解热镇痛药药物相互作用研究,所以是否同所有非雷体解热镇痛同时服用均会提高胃肠道出血事件还不清除。
因此,非幽体解热镇痛药和氯毗格雷同时口服时应小心。

氯吡格雷作用氯吡格雷是一种常见的抗血小板药物,常用于预防和治疗血栓性疾病。
它通过抑制血小板功能,防止血小板聚集和血栓形成,起到抗血栓的作用。
氯吡格雷主要通过作用于血小板上的腺苷二磷酸受体(ADP受体),抑制ADP与受体之间的结合。
这样一来,ADP不能激活血小板,血小板不能释放内源性凝血活性物质,从而减少血小板聚集和粘附,阻止了血栓的形成。
相比于其他抗血小板药物,氯吡格雷具有作用速度快、可逆性强、副作用少等优势,被广泛应用于临床。
在临床上,氯吡格雷主要用于治疗缺血性心血管疾病,如冠心病、心肌梗死和不稳定型心绞痛等。
在这些疾病中,血管内的血小板容易聚集和粘附,形成血栓,导致血管阻塞,引起血液循环不畅,加重疾病病情。
通过使用氯吡格雷可以有效地减少血小板聚集,预防血栓的形成,降低心血管事件的发生率,改善患者的生活质量。
氯吡格雷也可以用于预防与血小板聚集相关的其他疾病,如缺血性卒中、外周动脉疾病和血小板聚集性血小板减少性紫癜等。
此外,氯吡格雷还可以用于一些体外循环手术前后的抗血栓治疗,预防血小板聚集和血栓的形成,保证手术的顺利进行。
虽然氯吡格雷具有较好的治疗效果,但仍存在一定的副作用和禁忌症。
其中最常见的副作用是出血,特别是胃肠道出血。
患者在使用氯吡格雷期间应密切观察,一旦出现不明原因的胃肠道出血症状,应及时就医。
此外,氯吡格雷还可能引起过敏反应、骨髓抑制等不良反应。
对于哺乳期妇女、严重出血和出血性疾病患者以及正在接受抗血小板治疗的患者,应避免使用氯吡格雷。
总之,氯吡格雷是一种广泛应用于临床的抗血小板药物,通过抑制血小板功能,减少血小板聚集和血栓形成,预防和治疗血栓性疾病。
在使用氯吡格雷时,患者应注意剂量的准确使用,并密切观察可能出现的副作用和禁忌症,及时就医以保证治疗的安全和有效。
同时,与医生保持良好的沟通和协商,合理用药,对患者的疾病诊疗效果有着积极的作用。
氯吡格雷用于冠心病治疗的效果以及不良反应
氯吡格雷是一种抗血小板药物,主要用于预防冠心病患者的血栓形成和心血管事件的
发生。
它作为P2Y12受体拮抗剂,通过抑制血小板聚集来发挥其疗效。
氯吡格雷在临床上被广泛应用于冠心病患者治疗中,其预防血栓形成的疗效已被证实。
研究发现,与安慰剂相比,氯吡格雷能够有效地降低冠心病患者的心肌梗死、不稳定性心
绞痛和脑梗死的发生率。
同时,该药物还能够减少冠状动脉造影术后的血小板聚集率和出
现再狭窄的概率,从而促进患者的康复。
氯吡格雷的剂量一般为75mg/天,与阿司匹林一起使用能够取得更好的疗效。
尽管该
药物在冠心病治疗中具有显著的疗效,但是其在使用过程中也会产生一些不良反应。
常见
不良反应包括胃肠道症状、出血、血小板减少等。
在临床应用过程中,需要严格监测患者
的血小板计数和凝血指标,确保患者的用药安全。
在使用氯吡格雷的过程中,需要注意以下几点:
1. 患者在用药期间要定期检查血小板计数和凝血功能指标,发现异常必须停药。
2. 剂量和疗程应在医生的指导下使用,尤其对于老年患者、肾功能受损者和同时使
用其他药物的患者,应特别注意。
3. 长期使用氯吡格雷可能会导致血小板的耐药性增强,从而降低疗效和增加出血风险。
4. 患者在使用氯吡格雷期间应避免使用其他抗血小板药物,以免增加出血风险。
总之,氯吡格雷是冠心病治疗中一种有效的抗血小板药物,在使用时需要注意其在降
低风险的同时也会增加出血风险,应在医生的指导下使用。
氯吡格雷用于冠心病治疗的效果以及不良反应
氯吡格雷是一种血小板抑制剂,可以防止血小板聚集并减少血栓的形成,从而预防心
血管疾病发生和进展。
它常被用于冠心病的治疗。
氯吡格雷的主要疗效是通过抑制ADP受体的激活,从而阻止血小板的聚集和血栓的形成。
它可用于冠心病患者的治疗,包括稳定性心绞痛、非ST段抬高型心肌梗死和不稳定性心绞痛等病情。
已有多项临床研究证实,与单独使用阿司匹林相比,联合应用氯吡格雷和阿司匹林可
以显著降低冠心病患者的心脏事件和死亡率。
一项名为“CURE”的研究表明,对于非ST段抬高型心肌梗死患者,联合使用氯吡格雷和阿司匹林能够显著降低30天和一年的主要不良心脏事件。
氯吡格雷也存在一些不良反应和副作用。
常见的不良反应包括胃肠道反应,例如胃痛、恶心、呕吐和腹泻等。
这些症状通常是轻度和暂时的,但有时可能会导致较严重的胃黏膜
损伤如消化性溃疡和出血。
还有一些患者对氯吡格雷过敏,表现为皮疹、荨麻疹、呼吸困难以及严重的过敏反应。
这些情况应立即停止使用氯吡格雷并就医。
其他较少见的不良反应包括血小板减少、肝功能异常、出血倾向和头晕等。
需要注意
的是,由于氯吡格雷会影响血小板的聚集功能,因此在使用氯吡格雷期间患者易于出血。
如果患者需要进行手术或有其他出血风险的情况,应在使用氯吡格雷之前告知医生。
氯吡格雷在冠心病的治疗中,通过抑制血小板的聚集和减少血栓形成,可以降低心脏
事件和死亡率。
患者在使用氯吡格雷时应注意可能的不良反应和副作用,并定期复查相关
指标以确保疗效和安全性。
在使用氯吡格雷之前,患者应咨询医生并遵循医生的指导。
我现在氯吡格雷的一晶型已经做出来了,但产品都沾到了壁上,对于工业化很不好弄,请问各位大侠有没有做出来不沾壁的。
这个品种首先要拨到晶型2的专利,否则,即使申报成功上市,也会有一大堆问题。
我觉得,他的专利是可以拨的,其实并不具有新颖性(在丙酮里就很容易制得晶型2),只是没有牵头的。
国内申报的大部分是侵犯专利的晶型,当然国内企业市场也做不大,再着说了,原料药企业也不卖原料药,老外也就无法举证--侥幸心理,甚至有些人根本就不知道啥是晶型,以为合成了就行。
I晶型最大的问题是不稳定,这是隐患。
1、简介:硫酸氢氯吡格雷是法国赛诺菲圣德拉堡制药公司于1986年研究开发成功的新一代的血小板聚集抑制剂,商品名为波立维。
该产品于 1998年3月在美国首次上市,随后进入欧洲、北美、澳洲、新加坡等多国市场,并于2001年8月在中国上市。
2、医保情况:不属于医保药品3、适应症:预防和治疗因血小板高聚集状态引起的心、脑及其它动脉的循环障碍疾病。
4、上市情况:赛诺菲公司(晶型II):片剂,商品名为波利维;深圳信立泰(晶型I):片剂,商品名为泰嘉。
5、在注册情况:据SFDA公开信息,申报企业较多,均在审评中,晶型I一家。
6、项目特点:赛诺菲原料药为II晶型,进口独家;深圳信立泰为I晶型,国内独家。
由于II晶型专利未到期,本公司另辟蹊径,进口符合欧盟标准、资质齐全、无专利保护的晶型I,首家进口。
7、本品特点:本品为氯吡格雷的硫酸氢盐,稳定性好,对胃肠道刺激性小。
8、申报类别:6类9、合作方式:我公司提供合法原料及资质,贵方凭此申报制剂,我公司可提供委托研发、协助注册等相关服务。
公司名称:随州佳科医药化工有限公司联系人:叶问先生( 销售经理)手机:013872867025电话:************传真:************地址:湖北省随州市随州经济技术开发区塞纳左岸1018号硫酸氢氯吡格雷2型英文名:Clopidogrel Hydrogen sulfate英文化学名称:(aS)-a-(2-Chlorophenyl)-6,7-dihydrothieno[3,2-c]pyridine-5(4H)-acetic acid methyl ester Hydrogen sulfate 中文化学名称:S(+)-2-(2-氯苯基)-2-(4,5,6,7-四氢噻吩)[3,2,C]并吡啶-5)乙酸甲酯硫酸氢盐CAS号:135046-48-9(硫酸盐)分子式:C16H16ClNO2S•H2SO4分子量:419.90.理化常数:白色或类白色的结晶性粉末;无臭;本品在水、甲醇、乙醇或冰醋酸中溶解;在丙酮或氯仿中极微溶解;在醋酸乙酯中几乎不溶;在0.1mol/L 盐酸溶液中溶。
执业药师继续教育氯吡格雷说明书近年来,随着医学科技的不断进步,药物的种类与功能日益丰富。
对于执业药师来说,不断更新知识、学习新药物的使用方法和副作用成为了必修课。
而在众多药物中,氯吡格雷作为一种抗血小板药物,在心血管疾病的临床应用中备受关注。
本文将深入探讨氯吡格雷的药理作用、剂量使用和注意事项,帮助执业药师更好地掌握这一重要药物的使用要点。
一、氯吡格雷的药理作用氯吡格雷是一种嘌呤类ADP受体拮抗剂,通过抑制ADP(腺苷二磷酸)受体的激活而阻断血小板聚集,从而发挥抗血小板作用。
这种作用使其在预防心脏血管疾病方面具有重要意义,如稳定型心绞痛、急性冠脉综合征及急性心肌梗死等疾病的治疗中都有着重要的地位。
二、氯吡格雷的剂量使用和注意事项1.剂量使用根据患者的具体情况和诊断结果,氯吡格雷的剂量使用需谨慎确定。
一般情况下,对于急性冠脉综合征患者,可以首先口服氯吡格雷300mg的loading dose,然后以75mg/d的维持剂量进行长期治疗。
另外,对于非Q波型心肌梗死患者,首剂为300mg,继续使用75mg/d。
需要特别注意的是,氯吡格雷的剂量对于老年患者、肾功能不全、合并糖尿病患者等特殊人裙的使用需要进行个性化调整,执业药师应当对此有清晰的认识。
2.注意事项在使用氯吡格雷时,执业药师需要特别注意一些重要的事项。
氯吡格雷具有出血风险,因此需要密切监测患者的出血指标,如凝血酶原时间、国际标准化比值等。
氯吡格雷与其他药物的相互作用需要引起重视,尤其是抗凝药物、非甾体抗炎药等药物可能增加出血风险,执业药师需要及时提醒患者。
氯吡格雷还可能引起胃肠道不良反应,如胃溃疡、出血等,执业药师应当告知患者注意监测。
三、个人观点和理解作为一名执业药师,我对氯吡格雷这一抗血小板药物的使用有着深刻的认识与理解。
在实际工作中,我发现对于药物的了解不仅需要掌握其基本药理作用、剂量使用和注意事项,更需要考虑到患者的个体差异、合并症及用药安全,这是执业药师的职责所在。
www.6chem.com **********************.2019 六鉴化工咨询网(www.6chem.com)
1
氯吡格雷的概况 1.1 氯吡格雷的基本概况 分子式:C16H16ClNO2S 分子量:321.82 CAS号:113665-84-2 结构式:
1.2 氯吡格雷的用途 用途:抗凝血药,用于脑血栓等。 氯吡格雷是一种新型的噻吩并吡啶类衍生物。它通过选择性地与血小板表面腺苷酸环化酶耦联的ADP受体结合而不可逆地抑制血小板的聚集,可减少血管中血栓形成,与同属化合物噻氯匹定比,其作用强度和耐受性高而副作用较少,临床用于预防心肌梗死、中风或有外周动脉疾病史患者的动脉粥样硬化。目前的资料显示,作为全新型的ADP受体拮抗剂,氯吡格雷是一种安全、有效的血小板聚集抑制剂。它为动脉粥样硬化血栓性疾病的预防提供了全方位的对策,与环氧化酶抑制剂如阿司匹林联用能降低急性冠脉综合征的危险性,提供短期及长期的心脏保护作用。 www.6chem.com **********************.2019 六鉴化工咨询网(www.6chem.com)
2
1.3 氯吡格雷的药理毒理及药代动力学 【药理毒理】 药效学特性:氯吡格雷是一种血小板聚集抑制剂。ATC分类为:BO1AC/04。氯吡格雷选择性也抑制二磷酸腺苷(ADP)与它的血小板受体的结合及继发的ADP介导的糖蛋白GPlllb/llla复合物的活化,因此可抑制血小板聚集,氯吡格雷必须经生物转化才能抑制血小板的聚集,但是还没有分离出产生这种作用的活性代谢产物。除ADP外,氯吡格雷还能通过阻断由释放的ADP引起的血小板活化的扩增,抑制其它激动剂诱导的血小板聚集。氯吡格雷不能抑制磷酸二酯酶的活性。氯吡格雷通过不可逆地修饰血小板ADP受体起作用。暴露于氯吡格雷的血小板的寿命受到影响。而血小板正常功能的恢复速率同血小板的更新有关。从第一天起,每天重复给氯吡格雷75mg,抑制ADP诱导血小板聚集,抑制作用在3-7天达到稳态。在稳态,每天服用氯吡格雷75mg平均抑制水平维持中40%-60%,在治疗中止后一般约在5天内血小板聚集和出血时间逐渐回到基线。氯吡格雷的临床疗效来自于CAPRIE临床试验。该试验的入组病人有19,185个,为多个中心,多国家,随机双盲比较氯吡格雷(75mg/天)和阿司匹林(325mg/天)作用的平行临床研究。 随机入选的病人为:1)有近期心肌梗死史(35天内)。2)近期缺血性中风史(7天-6个月内),至少近一周内仍有继发神经系统症状。3)确诊外周动脉疾病(PAD)病人接受随机治疗1-3年,在心肌梗死组中,大多数患者在急性心肌梗死后的初期服用阿司匹林。 与阿匹林相比,氯吡格雷可显著降低新的缺血性事件(包括心肌梗死,缺血性中风和其它血管疾病死亡)的发生率。其中,939件发生在氯吡格雷治疗组,1020件发生在阿司匹林治疗组(相对危险降低(RRR)8.7%[95% CI:0.2-16.4];P=0.045),相当于每1000名病人接受2年治疗,10[CI:0-20]名病人就可避免出现一次缺血性事件。氯吡格雷治疗组和阿司匹林治疗组的总体死亡率分别为5.8%和6.0%,没有显著性差异。根据心肌梗死,缺血性中风和其它血管疾病死亡进行分组分析,由于PAD(尤其是那些有心肌梗死史的病人)(RRR=23.7%;CI:8.9-36.2)入组的病人和由于 www.6chem.com **********************.2019 六鉴化工咨询网(www.6chem.com)
3
严重缺血性中风(与阿司匹林治疗组相比没有显著性差异)(RRR=7.3%;CI-5.7-18.7)入组的病人受益最大(p=0.003)。 由于近期心肌梗死而入选的病人,氯吡格雷治疗组的有效率略低于阿司匹林治疗组,但在统计学上无差异(RRR=4.0%;CI:-22.5-11.7)。而且,根据年龄分组分析,氯吡格雷对75岁以下病人的治疗作用好于75岁以上病人。由于CAPRIE临床试验并没刻意设计来评价氯吡格雷对某组病人更有效,所以这种差异是否真实或是偶然还是不清楚。 临床前安全性研究大鼠和狒狒临床前最常见的反应为肝脏发生变化。所服剂量为人体服用75mg/天氯吡格雷后使用剂量的25倍,这些肝脏变化是由于药品对肝脏代谢酶影响的结果。大鼠和狒狒服用高剂量氯吡格雷,胃耐受性差(胃炎、胃溃疡和/或眩晕)。以每天大至77mg/kg剂量,小鼠服用78周,大鼠服用104周的氯吡格雷没有发现致癌的证据。此剂量的血药浓度较人类的推荐剂量(每天75mg)大25倍。经过一系列体内的体外试验证实氯吡格雷无致突变效果。氯吡格雷对雌性大鼠和雄性大鼠的生育能力没有影响,对大鼠和兔子均无致畸作用。哺乳大鼠服用氯吡格雷可轻微延缓幼儿的发育。药代动力学研究表明氯吡格雷和/或其代谢物从乳汁中排泄,因此,不排除氯吡格雷有直接(轻微毒性)或间接(味道不好)作用。 【药代动力学】 多次口服氯吡格雷75mg以后,氯吡格雷吸收迅速,母体化合物的血浆浓度很低,一般在用药2小时后低于定量限(0.00025mg/L)。根据尿液中氯吡格雷代谢物排泄量计算,至少有50%的药物被吸收。 氯吡格雷主要由肝脏代谢。血中主要代谢产物是羧酸盐衍生物,其对血小板聚集也无影响,占血浆中药物相关化合物的85%。多次口服氯吡格雷75mg以后,血药浓度约在1小时后达峰(30mg/l) 氯吡格雷主要由一种药物前体,通过氧化作用形成2-氧基-氯吡格雷,然后再经过水解形成活性代谢物(一种硫醇衍生物)。氧化作用主要由细胞色素P450同功酶2B6和3A4调节,1A1、1A2和2C19也有一定的调节作用。体外分离这种活性代谢物显示它可迅速不可逆的与血小板受体结合,从而抑制血小板聚集。但在血中未 www.6chem.com **********************.2019 六鉴化工咨询网(www.6chem.com)
4
检测到此种代谢物。 在氯吡格雷50-150mg范围内,主要代谢物药代动力学为线性增长(血浆浓度与剂量成正比)。 在很广的浓度范围内,氯吡格雷及其主要代谢物均可在体外与人体的血浆蛋白可逆性结合(分别为98%和94%)。 人体口服14C标记的氯吡格雷以后,在5天内约50%由尿液排出,约46%由粪便排出,一次和重复给药后,血浆中主要代谢产物的消除半衰期为8小时。 每天重复服用波立维75mg后,严重肾损害病人(肌酐清除率5-15ml/min)和健康志愿者。尽管ADP诱导血小板聚集抑制作用低于健康志愿者25%,但出血时间的延长与每天服用氯吡格雷75mg的健康志愿者相同。而且,所有病人的临床耐受性良好。 健康志愿者及患有肝硬化(Child-Pugh classA或B)病人单次,多剂量服用氯吡格雷,对氯吡格雷药效学硬化病人单次,多剂量服用氯吡格雷,对氯吡格雷药效学及药代动力学进行研究。表明每天一次服用氯吡格雷75mg,进行10天,药物安全,受试者对药物耐受良好。肝硬化病人单次服药及稳态氯吡格雷血药浓度峰值高于健康志愿者几倍。但肝硬化组和健康志愿者组间血中主要代谢物浓度,结ADP诱导血小板聚集的抑制作用和出血时间均相当。
1.4 氯吡格雷的用法用量及不良反应
【适应症】 适用于有过近期发作的中风、心肌梗死和确诊外周动脉疾病的患者。该药可减少动脉粥样硬化性事件的发生(如心肌梗死,中风和血管性死亡。) 【用法用量】 推荐剂量为每天 75mg,与或不与食物同服,对于老年患者不需调整剂量。 【不良反应】 通过对11,300多病人的治疗,其中7000多病人接受治疗1年或以上,评价氯吡格雷的安全性。大型临床研究(CAPRIE)中,服用75mg/天氯吡格雷,与服用325mg/ www.6chem.com **********************.2019 六鉴化工咨询网(www.6chem.com)
5
天阿司匹林相比耐受良好。不论年龄,性别和种族,氯吡格雷的总体耐受性与阿司匹林类似。在CAPRIE试验中临床主要的不良反应讨论如下: 出血: 接受氯吡格雷或阿司匹林治疗的病人,出血的总发生率为9.3%。氯吡格雷和阿司匹林严重出血事件的发生率分别为1.4%和1.6%。 接受氟吡格雷治疗的病人,胃肠道出血的发生率为2.0%,需住院治疗的为0.7%,而阿司匹林分别为2.7%和1.1%。 与阿司匹林相比,服用氯吡格雷的病人其他出血事件的发生率高(7.3%vs6.5%),但两个治疗组的严重事件发生率相似(0.6%vs0.4%).两个治疗组最常见不良事件为:紫癜/挫伤/血肿,和鼻出血,其他还有血肿、血尿和眼部出血(主要是结膜出血)。 颅内出血发生率氯吡格雷为0.4%,阿司匹林为0.5%。 血液病: 有6个病人出现严重中性白细胞减少症(中性白细胞﹤0.45×109/1),4个属于氯吡格雷组( 0.04%),2个属于阿司匹林组(0.02%)。9599个氯吡格雷组病人中有两人出现中性白细胞数为零,而阿司匹林组的9586个病人中无人出现。氯吡格雷组病人中出现一例再生障碍性贫血。 氯吡格雷组严重血小板减少症(﹤80×109/1)发生率为0.2%,阿司匹林组为0.1%;出现血小板计数≦30×109/1的情况非常少。 胃肠道: 总体来讲,胃肠道反应的发生率(如腹痛、消化不良、胃炎和便秘)氯吡格雷组为27.1%,而阿司匹林组为29.8%。而且,由于胃肠道的副作用而退出治疗的氯吡格雷组为3.2%,阿司匹林组为4.0%。但是,各组临床严重副反应的发生率没有统计学差异(3.0%vs.3.6%)。两个治疗组最常见不良事件为:腹痛、消化不良、腹泻和恶心。其他还有便秘、牙病症、眩晕和胃炎等。 腹泻发生率氯吡格雷组为4.5%,明显高于阿司匹林组(3.4%)。严重腹泻的发生率两治疗组相似(0.2%vs.0.1%)。消化道,胃及十二指肠溃疡的发生率氯吡格雷组为0.7%,而阿司匹林组为1.2%。