第三章 第二节 药动学基本参数及概念
- 格式:doc
- 大小:175.50 KB
- 文档页数:6


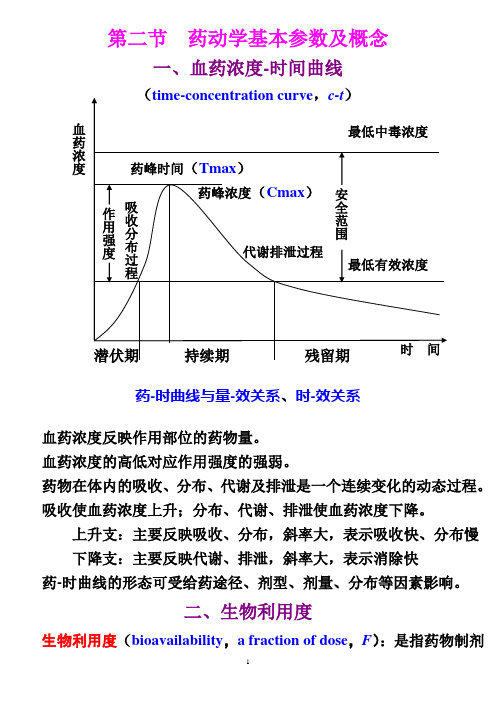
第二节 药动学基本参数及概念一、血药浓度-时间曲线药-时曲线与量-效关系、时-效关系血药浓度反映作用部位的药物量。
血药浓度的高低对应作用强度的强弱。
药物在体内的吸收、分布、代谢及排泄是一个连续变化的动态过程。
吸收使血药浓度上升;分布、代谢、排泄使血药浓度下降。
上升支:主要反映吸收、分布,斜率大,表示吸收快、分布慢 下降支:主要反映代谢、排泄,斜率大,表示消除快药-时曲线的形态可受给药途径、剂型、剂量、分布等因素影响。
二、生物利用度生物利用度(bioavailability ,a fraction of dose ,F ):是指药物制剂血药浓度被机体吸收的速率和吸收程度的一种量度。
即:实际被吸收利用的量(A)占服用总量(D)的百分比。
F=A/D×100%药物的吸收量可通过测定给药后的药-时曲线下面积(area under the time concentration curve,AUC)来估算。
绝对生物利用度F=AUC po×D iv/AUC iv×D po×100%相对生物利用度F=AUC t×D r/AUC r×D t×100%AUC:曲线下面积D:剂量po:口服iv:静注t:试验制剂r:参比制剂影响因素:可因制剂质量、剂型、给药途径、患者具体情况等不同,同一药物的生物利用度会有差异。
意义:评价各种药物制剂的生物等效性;评价药物的首过消除与作用强度;指导临床合理用药;查明药物无效或中毒的原因。
三、表观分布容积药物在体内的分布是不均的。
当药物在体内分布达到动态平衡时,体内药量与血药浓度的比值称表观分布容积(apparent volume of distribution,V d)。
V d=A/C。
V d:表观分布容积A:给药量(mg/kg)C:血药浓度(mg/L)意义:表示药物在组织中分布范围的广窄,结合程度的高低。
V d≈0.045 L/kg,主要分布在血浆;V d= 0.14-0.29 L/kg,主要分布在细胞外液;V d= 0.3-0.4 L/kg,主要分布在细胞内液;V d≈0.6L/kg,表示分布在细胞内外液;V d>>0.6L/kg,表示药物在某组织中集中分布。


生物药剂学与药动学——药动学概述一、药动学定义药动学是应用动力学的原理和数学处理方法,研究药物在体内的吸收、分布、代谢和排泄过程(即ADME 过程)的量变规律的学科,即药动学是研究药物体内过程动态变化规律的一门学科。
二、血药浓度与药物效应(一)治疗浓度范围治疗浓度范围即治疗窗,是指给药后产生药效的最低有效浓度和产生毒性的最低中毒浓度之间的浓度范围。
治疗窗窄的药物,其治疗浓度相对较难控制,易发生治疗失败或不良反应,常需进行治疗药物监测。
(二)血药浓度与药物效应的关系对于大多数药物及其制剂,药物进入体内后,血中的药物浓度与药物作用靶位的实际浓度呈正相关,从而间接反映药物的临床效应,包括治疗效果及不良反应。
药动学中常以血液中的药物总浓度作为观察指标。
三、药动学的基本概念和主要参数(一)血药浓度-时间曲线药动学的研究中,将药物制剂通过适当的方式给予受试者,然后按照适当的时间间隔抽取血样,检测血样中的药物浓度,每一个取血时间点有一个对应的药物浓度,由此就得到一系列的血药浓度相对于时间的实验数据,简称为药-时数据。
将其用坐标图表示,称为血药浓度-时间曲线,简称药-时曲线。
血管内给药的药-时曲线通常为曲线,而血管外给药的药-时曲线一般为拋物线。
根据研究的需要,常将药-时曲线的不同时间段用吸收相、平衡相和消除相来表示,表明该时间段(时相)体内过程的主要影响。
(二)血药浓度-时间曲线下面积血药浓度-时间曲线图中,药-时曲线与时间轴共同围成的面积称为血药浓度-时间曲线下面积,简称药-时曲线下面积,用AUC表示。
其与药物吸收的总量成正比,能够反映药物吸收的程度。
AUC越大,表明制剂中的药物被生物体吸收越完全。
血药浓度-时间曲线下面积是评价制剂生物利用度和生物等效性的重要参数。
(三)峰浓度和达峰时间血管外给药的药-时曲线一般为拋物线,其中有两项特征性参数,即血药峰浓度和达峰时间。
血药峰浓度即药-时数据中的最大浓度,用C max表示,C max的大小能够反映药物的疗效情况和毒性水平。

药动学掌握药物的吸收、分布及其影响因素,P450酶系及其抑制剂和诱导剂,药物排泄途径及其影响肾排泄的因素,血浆蛋白结合率和肝肠循环的概念。
药物代谢动力学,简称为药动学,研究药物体内过程及体内药物浓度随时间变化的规律。
药物在体内分布达到平衡后药理效应强弱与药物血浆浓度成比例。
医生可用药动学规律计算药物剂量以达到所需的血药浓度并掌握药效的强弱久暂。
比单凭经验处方取得较好的疗效。
第一节药物体内过程一、药物的跨膜转运药物在体内的过程:吸收、分布、生物转化、排泄,需进行跨膜转运的过程是吸收、分布、排泄。
1、被动转运(顺梯度转运): 药物依赖于膜两侧的浓度差,从高浓度的一侧向低浓度的一侧扩散转运的过程。
多数药物属于被动转运。
(1)特点:不需要载体,不消耗能量,无饱和现象和竞争性抑制。
(2)影响扩散速度的因素:①膜两侧的药物浓度差。
②药物理化性质:分子量小、脂溶性大、极性小、非解离型的药易通过生物膜转运,反之难跨膜转运。
2、主动转运:是一种逆浓度(或电位)差的转运。
特点:需要载体,消耗能量,有饱和现象和竞争性抑制。
二、吸收药物的吸收是指药物进入血液循环的过程。
静脉注射无吸收过程。
吸收速度与程度主要取决于药物的理化性质、剂型、剂量和给药途径。
(一)吸收方式1.多数药按简单扩散进入(吸收)。
(1)影响扩散速度的因素:1)膜的性质,面积及膜两侧的浓度梯度,2)药物的性质,分子量小的(200D以下),脂溶性大的(油水分布系数大的),极性小的(不易离子化的)药较易通过。
(2)吸收分布排泄的一个可变因素,与环境的酸碱度有关。
(3)离子障现象:非离子型药可自由穿透,而离子型药被限制在膜的一侧。
离子障与吸收有关,可以理解为“酸酸易吸收,酸碱难吸收”。
如弱酸性药在胃液中非离子型多,在胃中即可被吸收。
弱碱性药在酸性胃液中离子型多,主要在小肠吸收。
2.少数药按主动转运而吸收,特点:1)与正常代谢物相似的药物,如5-氟尿嘧啶、甲基多巴等;2)靠载体主动转运而吸收的;3)对药物在体内分布及肾排泄关系密切。
药动学概述学习要点:1.药动学基本参数及其临床意义2.房室模型:单室模型、双室模型、多剂量给药3.非线性动力学4.给药方案设计5.个体化给药6.治疗药物监测7.新药药动学研究8.生物利用度9.生物等效性药物动力学(药物代谢动力学、药代动力学)——研究药物在体内的动态变化规律,定量描述需要搞懂药动学的三大人群新药研发临床试验临床药师一、药动学基本概念1.血药浓度-时间曲线(药时曲线)药动学的研究中,将药物制剂通过适当的方式给予受试者,然后按照适当的时间间隔抽取血样,检测血样中的药物浓度,每一个取血时间点有一个对应的药物浓度,由此就得到一系列的血药浓度相对于时间的实验数据,简称为药-时数据。
将其用坐标图表示,称为血药浓度-时间曲线(药-时曲线)2.治疗浓度范围(治疗窗)治疗窗窄的药物,其治疗浓度相对较难控制,易发生治疗失败或不良反应,常需进行治疗药物监测。
3.血药浓度与药物效应的关系大多数药物进入体内后,血中的药物浓度与药物作用靶位的实际浓度呈正相关,从而间接反映药物的临床效应,包括治疗效果及不良反应。
部分药物在血液中可能与血浆蛋白结合,药物的存在形式包括结合型与游离型,只有游离型药物能通过生物膜到达作用部位。
血液中的游离型药物浓度常与总浓度保持一定的比例,药动学中常以血液中的药物总浓度作为观察指标。
4.药物转运的速度过程①一级速度过程速度与药量或血药浓度成正比。
②零级速度过程速度恒定,与血药浓度无关恒速静滴给药速度、控释制剂药物释放速度、酶饱和后转运③受酶活力限制的速度过程(Michaelis-Menten型、米氏方程)浓度影响反应速度,药物浓度高出现酶活力饱和。
高浓度零级,低浓度一级5.药动学常用参数药动学参数计算含义速率常数k(h-1、min-1)吸收:k a消除k=k b+k e+k bi+k lu…速度与浓度的关系,体内过程快慢生物半衰期(t1/2)t1/2 =0.693/k消除快慢——线性不因剂型、途径、剂量而改变,半衰期短需频繁给药表观分布容积(V)V=X0/C0表示分布特性——亲脂性药物,血液中浓度低,组织摄取多,分布广(地高辛vs利福平)清除率Cl=kV 消除快慢A:关于药动力学参数说法,错误的是A.消除速率常数越大,药物体内的消除越快B.生物半衰期短的药物,从体内消除较快C.符合线性动力学特征的药物,静脉注射时,不同剂量下生物半衰期相同D.水溶性或者极性大的药物,溶解度好,因此血药浓度高,表观分布容积大E.清除率是指单位时间内从体内消除的含药血浆体积『正确答案』DA:地高辛的表观分布容积为580L,远大于人体体液容积,原因可能是A.药物全部分布在血液B.药物全部与血浆蛋白结合C.大部分与血浆蛋白结合,与组织蛋白结合少D.大部分与组织蛋白结合,药物主要分布在组织E.药物在组织和血浆分布『正确答案』DA:某药物按一级速率过程消除,消除速率常数k=0.095h-1,则该药物消除半衰期t1/2约为A.8.0hB.7.3hC.5.5hD.4.0hE.3.7h『正确答案』BA:静脉注射某药,X0=60mg,若初始血药浓度为15μg/ml,其表观分布容积V是A.0.25LB.2.5LC.4LD.15LE.40L『正确答案』CA.0.2303B.0.3465C.2.0D.3.072E.8.42给某患者静脉注射一单室模型药物,剂量为100.0mg,测得不同时刻血药浓度数据如下表。
药动学的基本参数及概念掌握药动学基本概念及其重要参数之间的相互关系:药-时曲线下面积、生物利用度、药峰时间、药峰浓度、消除半衰期、表观分布容积、清除率等。
一、一次性血管外给药三个时期1、潜伏期(短:吸收快)——有效期——残留期(长:蓄积中毒)2、时量关系:血药浓度随时间的变化过程。
3、房室概念与房室模型1)一室模型:假定身体由一个房室组成,给药后药物立即均匀地分别于整个房室,并以一定的速率从该室消除。
单次静注给药时,时量(对数浓度)曲线呈单指数消除。
2)二室模型:假定身体由两个房室组成,即中央室(血流丰富的器官如心、肝、肾)和周边室(血流量少的器官如骨、脂肪)。
给药后药物立即分布到中央室,然后缓慢分布到周边室。
单次静注给药时,时量(对数浓度)曲线呈双指数衰减即分为分布相和消除相。
二、药动学重要参数1、消除半衰期及意义:血药浓度下降一半所需的时间。
是决定给药间隔时间的重要参数之一。
2、生物利用度:药物吸收速度与程度的一种量度。
可药时曲线下面积AUC计算,F=口服AUC/注射AUC。
3、表观分布容积Vd :是指血药浓度与体内药物量间的一个比值,Vd=A/C=体内药量/血药浓度。
可反映药物分布的广泛程度或药物与组织结合的程度。
4、药-时曲线下面积AUC 代表一次用药后的吸收总量,反映药物的吸收程度。
三、药物消除动力学1、一级消除动力学(恒比消除):多数药消除半衰期恒定,与血药浓度无关。
血浆清除率(Cl):即单位时间内多少容积血浆中的药物被消除干净(单位用L•h-1 )。
消除速率:单位时间内被机体消除的药量。
常用表观分布容积(Vd)计算。
Vd及Cl的区别:①是两个独立的药动学指标,各有其固定的数值,互不影响,也不因剂量大小而改变其数值。
②Vd是表观数值,不是实际的体液间隔大小。
多数药的Vd值均大于血浆容积。
③Cl不是药物的实际排泄量。
Cl是肝肾等消除能力的总和。
④与组织亲和力大的脂溶性药物其 Vd可能比实际体重的容积还大。
矿产资源开发利用方案编写内容要求及审查大纲
矿产资源开发利用方案编写内容要求及《矿产资源开发利用方案》审查大纲一、概述
㈠矿区位置、隶属关系和企业性质。
如为改扩建矿山, 应说明矿山现状、
特点及存在的主要问题。
㈡编制依据
(1简述项目前期工作进展情况及与有关方面对项目的意向性协议情况。
(2 列出开发利用方案编制所依据的主要基础性资料的名称。
如经储量管理部门认定的矿区地质勘探报告、选矿试验报告、加工利用试验报告、工程地质初评资料、矿区水文资料和供水资料等。
对改、扩建矿山应有生产实际资料, 如矿山总平面现状图、矿床开拓系统图、采场现状图和主要采选设备清单等。
二、矿产品需求现状和预测
㈠该矿产在国内需求情况和市场供应情况
1、矿产品现状及加工利用趋向。
2、国内近、远期的需求量及主要销向预测。
㈡产品价格分析
1、国内矿产品价格现状。
2、矿产品价格稳定性及变化趋势。
三、矿产资源概况
㈠矿区总体概况
1、矿区总体规划情况。
2、矿区矿产资源概况。
3、该设计与矿区总体开发的关系。
㈡该设计项目的资源概况
1、矿床地质及构造特征。
2、矿床开采技术条件及水文地质条件。