双原子分子电子态振动跃迁F-C因子的计算
- 格式:doc
- 大小:528.00 KB
- 文档页数:9






第一章 绪 论1. 解释下列名词:(1)仪器分析和化学分析;(2)标准曲线与线性范围;(3)灵敏度、精密度、准确度和检出限。
答:(1)仪器分析和化学分析:以物质的物理性质和物理化学性质(光、电、热、磁等)为基础的分析方法,这类方法一般需要特殊的仪器,又称为仪器分析法;化学分析是以物质化学反应为基础的分析方法。
(2)标准曲线与线性范围:标准曲线是被测物质的浓度或含量与仪器响应信号的关系曲线;标准曲线的直线部分所对应的被测物质浓度(或含量)的范围称为该方法的线性范围。
(3)灵敏度、精密度、准确度和检出限:物质单位浓度或单位质量的变化引起响应信号值变化的程度,称为方法的灵敏度;精密度是指使用同一方法,对同一试样进行多次测定所得测定结果的一致程度;试样含量的测定值与试样含量的真实值(或标准值)相符合的程度称为准确度;某一方法在给定的置信水平上可以检出被测物质的最小浓度或最小质量,称为这种方法对该物质的检出限。
2. 对试样中某一成分进行5次测定,所得测定结果(单位μg ⋅mL -1)分别为 0.36,0.38,0.35,0.37,0.39。
(1) 计算测定结果的相对标准偏差;(2) 如果试样中该成分的真实含量是0.38 μg ⋅mL -1,试计算测定结果的相对误差。
解:(1)测定结果的平均值37.0539.037.035.038.036.0=++++=x μg ⋅mL -1标准偏差122222120158.015)37.039.0()37.037.0()37.035.0()37.038.0()37.036.0(1)(-=⋅=--+-+-+-+-=--=∑mL g n x x s ni iμ相对标准偏差 %27.4%10037.00158.0%100=⨯=⨯=x s s r(2)相对误差 %63.2%10038.038.037.0%100-=⨯-=⨯-=μμx E r 。
3. 用次甲基蓝-二氯乙烷光度法测定试样中硼时,为制作标准曲线,配制一系列质量浓度ρB (单位mg ⋅L -1)分别为0.5,1.0,2.0,3.0,4.0,5.0的标准溶液,测得吸光度A 分别为0.140,0.160,0.280,0.380,0.410,0.540。



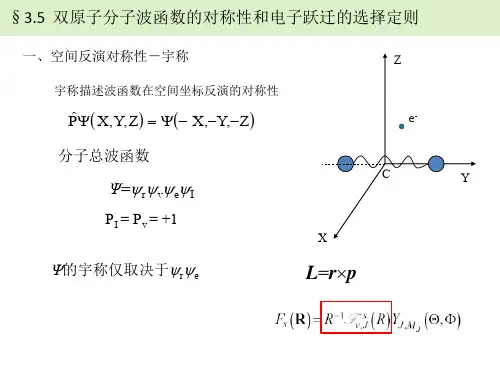
双原子分子结构、填空题(在题中空格处填上正确答案)3101、描述分子中________________ 空间运动状态的波函数称为分子轨道。
3102、在极性分子AB中的一个分子轨道上运动的电子,在A原子的A原子轨道上出现的概率为80%, B原子的B原子轨道上出现的概率为20%,则该分子轨道波函数。
3103、设A和B分别是两个不同原子A和B的原子轨道,其对应的原子轨道能量为E A和E B,如果两者满足___________ ,_______________ , _______ 原则可线性组合成分子轨道=C A A + C B B。
对于成键轨道,如果E A_________ E B,贝U C A _____C B。
(注:后二个空只需填"=",">"或”等比较符号)3104、试以Z轴为键轴,说明下列各对原子轨道间能否有效地组成分子轨道,若可能,则填写是什么类型的分子轨道。
3105、判断下列轨道间沿z轴方向能否成键。
如能成键,则在相应位置上填上分子轨道的名称。
3106、AB为异核双原子分子,若A dyz与B P可形成型分子轨道,那么分子的键轴yzy编辑版word为____ 轴。
3107、若双原子分子AB的键轴是z轴,则A的d yz与B的P y可形成_________________ 型分子轨道。
3108、以z轴为键轴,按对称性匹配原则,下列原子轨道对间能否组成分子轨道?若能,写出是什么类型分子轨道,若不能,写出”不能",空白者按未答处理。
________________ ,磁性 _________________ 。
3110、在z方向上能与d xy轨道成键的角量子数l w 2的原子轨道是 _______________ 形成的分子轨道是___________ 轨道。
3111、在x方向上能与d xy轨道成键的角量子数K 2的原子轨道是__________________ 3112、用分子轨道表示方法写出下列分子基态时价层的电子组态:N2:_______________________________ ,02: ____________________________ 。
二、多原子分子的振动态正则振动一个由N个原子组成的分子的核运动有3N个自由度。
分子的核运动:分子整体的平动:需要3个自由度描述;非线形分子的转动:需要3个自由度描述;线形分子的转动:需要2个自由度描述;分子的振动自由度:非线形分子是s= 3N-6线形分子是s= 3N-5 双原子分子有3×2-5=1个振动自由度选择适当的坐标系,使得动能和势能中的交叉项消失(即i ≠j时,a ij= 0, b ij= 0, )。
即找到一个变换矩阵R,把原坐标(q1, q2, q3,⋯,qs)变换到新的坐标(Q1, Q2, …, Qs)1122s sq Qq QRq Q⎛⎫⎛⎫⎪ ⎪⎪ ⎪=⎪ ⎪⎪ ⎪⎝⎭⎝⎭111212122212sss s ssr r rr r rRr r r⎛⎫⎪⎪=⎪⎪⎝⎭其中,例如:CO 2是非中心对称的线形三原子分子,有4个振动自由度,所以有四种简振模式:(a) 对称伸缩振动=1285.8 cm -1~ν1(b) 反对称伸缩振动=2350.1 cm -12ν C O O C O O(c) (d) 弯曲振动= 677.5 cm -134νν= yxz x z (c)y x z x z y y (d)显然,(a)模式不改变分子的电偶极矩(等于零),所以在红外振动光谱上观测不到;(b)(c)(d)模式则会改变分子的正负电荷中心,引起电偶极矩的变化,所以可以在红外振动光谱上观测到。
又如:H 2O 属C 2v 点群,是非线形三原子分子,有3个振动自由度,所以有3中简正振动模式:(a) 对称伸缩振动=3600 cm -1~ν1(b) 对称弯曲振动= 1595.4 cm -12ν(c) 反对称伸缩振动= 3758.35 cm -13ν在谐振子模型下,在同一电子态上涉及两个不同的简正振动模式i 和j 的振动能级之间的(红外)光学跃迁,选择定择则为11,2,,i v i s ∆=±= 0j v j i∆=≠如考虑到振动的非谐性,则∆v i 可取任意整数,即一个简正振动模式可以在跃迁中改变好几个量子,也可以同时在几个简正振动模式中改变几个量子,这样的谱带称为泛频谱带,其强度比基频谱带( )弱。
双原子分子电子态振动跃迁F-C因子的计算摘要:分子激光冷却作为当前冷分子研究的热点,受到广泛关注。
针对激光冷却分子中如何找到合适的候选分子,最为关键的问题就是要找到这些分子部的结构参数、能量光谱,从而为下一步的能级激发奠定基本依据。
本文结合F-C因子计算原理,给出了F-C因子计算的理论解释;以MgF双原子分子为例,提出一种利用Morse 精确求解薛定谔方程;通过计算,得到MgF不同振动态的F-C 因子。
通过利用这种方法,也为激光冷却实验中的光谱数据测量提供了参考。
关键词:振动跃迁;Franck - Condon 因子;振动结构强度;Morse势能函数Calculation of the F-C factor of the electronic states of the doubleatom moleculeAbstract: molecular laser cooling, as a hot spot in the research of cold molecules, has received extensive attention. In order to find a suitable candidate molecule in laser cooling, the key problem is to find the molecular structure parameters, the internal energy spectrum, which lay a fundamental basis for the next step of the excitation level. This paper combines the F-C factor calculation principle, explain the F-C factor calculation theory; secondly, the MgF diatomic molecule as an example, using a Morse exact solution of Schrodinger equation; finally through calculation, MgF dynamic different vibration factor F-C. By using this method, it also provides a reference for the measurement of spectral data in laser cooling experiments.Key words: vibrational transition; Condon - Franck factor; vibrational structure intensity; Morse potential energy function在分子结构与分子光谱理论中,有关分子光谱带系的跃迁几率、谱线相对强度等光谱参量的计算公式都包含 Franck - Condon 因子(简称 F-C 因子) , 可以说 , F- C 因子与分子光谱问题的研究直接相关. 双原子分子 F- C 因子的计算在化学物理、定量光谱学 、等离子体辐射诊断等领域中都有着极其重要的意义. 计算F- C 因子,常常需要应用各种数值计算方法来求解能量算符的本征值方程。
本文则结合F-C 原理,提出一种通过 Morse 对薛定谔方程进行求解,从而得到其振动波函数。
1、Franck-Condon 原理1925 年 Franck 在分析光解离现象,讨论分子光解离过程时,提出了一套假说。
他认为:在“冷”气体中,分子无振动地处于最低电子态(基态),原子核的运动由势函数)(1r V 决定,如果分子吸收一份量子的能量能够从基态跃迁到激发态,那么在这样的过程中,除分子电子态的能量发生变化外,无其它变化。
光的吸收仅仅使原子核的相互作用势由)(1r V 变为)(2r V (即)(2r V 代替)(1r V ),但由于新的作用势)(2r V 有不同于)(2r V 的平衡位置,分子吸收光能量后,原子将离开平衡位置开始振动。
如果max 2012)(V r V >(其中,max 2V 是)(2r V 在区域 ∞<<r r 01中的最大值),分子趋于无限大振幅振动,分子发生解离。
1926 年 Condon 发展了 Franck 的假说,将上述观点推广到分子的电子跃迁,不管是吸收还是发射过程,以及初始状态有无振动。
主要观点是:(1)由于原子核的质量远远大于电子的质量,相对于原子核周期性振动而言,电子跃迁被认为是发生在可以忽略的极短时间。
(2)如果跃迁时刻的原子间距是r ,动量是r p ,那么可以假设电子跃迁不改变r 和r p 值,仅仅以一个新的势函数代替原先的势函数。
(3)电子跃迁时刻的r 和r p 值,完全决定末态分子的振动运动。
两年后,Condon 又用全量子理论对上述观点进行了解释。
1.1电子谱带振动结构强度分布的经典解释Franck-Condon 原理可以解释分子电子光谱振动结构的强度分布。
由于分子中的电子跃迁发生得很快(约 10-16秒),而核运动的周期较大(约 10-13秒),以至跃迁前后的核间距和核运动速度几乎不变,换句话说,在两个势能曲线之间垂直向上和向下跃迁的几率最大。
也就说是在发生电子跃迁时,分子中各原子核的位置及其环境可视为几乎不变。
即垂直跃迁的几率最大。
这个思想是 J. Franck 在 1925年首先提出来的。
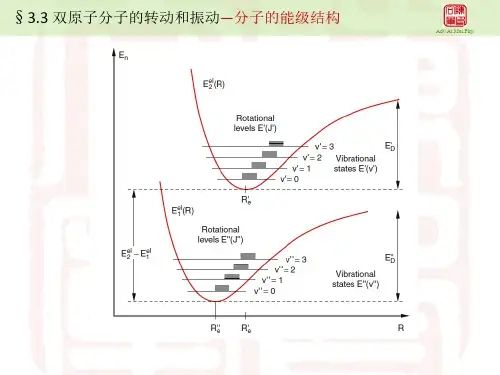
图1 对吸收光谱的解释对于吸收光谱,因为大多数分子原来都处于基态,即如图1中,势能曲线e E 的最低点 A 处(忽略零点振动)。
根据 Franck-Condon 原理,电子跃迁后的一瞬间,分子将处于 A 点正上方上势能曲线'e E 上的 B 点处。
B 点处对应的分子核间距和 A 点处的相同,并且相对速度为零(势能曲线上各点的振动动能为零)。
根据两势能曲线的最低点核间距 R 值的对比,可以解释吸收光谱振动带强度分布的不同情况。
1.2电子谱带振动结构强度分布的量子力学解释1928 年 E.Condon 运用波动力学,进一步解释了 Franck-Condon 原理。
我们知道,发射谱或吸收谱的强度与电偶极跃迁矩平方2mn d 成正比,mn d 是一个积分 :⎰*==τψψd d n d m d mn ˆ|ˆ|(1)其中*ψ和ψ是相结合的两个态的总波函数,dˆ是电偶极算符。
在考虑振动结构强度时可不涉及转动波函数,所以总波函数近似为电子与振动波函数乘积:v e Eev φφψ==|(2)另一方面分子的电偶极矩d ˆ可以分为核的电偶极矩nd ˆ与电子的电偶极矩ed ˆ: d ˆ=n d ˆ+ed ˆ(3) 因此,根据上述的分析,我们可以将dˆ的矩阵元分为两项: ⎰⎰**==ee e v e e mn dV vdR v E d v E d φφφφ''|ˆ|''(4) 在上述公式中,'v φ和v φ表示不同电子态的振动波函数。
谱带强度与公式(4)右边的重叠积分的平方成正比,有:[]2'dR I v v φφ*∝(5)而公式(5)中的[]2'dR v v φφ*则表示为F-C 因子。
2、F-C 因子计算基于 Franck-Condon 原理,我们将振动谱带的跃迁强度的量子表达公式为:***=∝⎰''22''2')(v v e v v e v v q R dR R I ψψ(6) 式中 *v v I '表示跃迁强度,*''v v ψψ、分别是上下两个电子态的振动波函数,ν′ 和 ν″ 表示上下两个态的振动量子数,用平均值 e R 2替换电子跃迁距e R , R 表示核间距,*''v v q 表示上下电子态的振动波函数的重叠积分的平方,即为 Franck-Condon 因子。
而根据Franck-Condon 因子的不同理论解释,可以将其对应的跃迁强度表示为:2''''''''')(⎰∝dR I j v j v v v ψψ(7)''''''j v j v ψψ、是振转波函数。
根据求解径向波函数vj vj vj vj E j R U dR d ψψψ=+-),(2122,其中J 为转动量子数, E 为本征能量,由此通过公式(7)可以得出近似波函数。
这样,在这里仅讨论无转动的情况下,要求出波函数vj ψ就得先得到势能函数),(J R U 。
通常可以用 Morse 势 )]}R - ex p[-βx p - {1D = U(R)2e e 代替势能函数,其中 e D 表示解离能,β 为 Morse 势能参数。
相较于Rees -Klein -Rydberg 法(RKR)或者是DPR 方法计算 F-C ,在近平衡状态下,用 Morse 势计算较为方便,而且与实验所测得值也比较吻合。
采用 Morse 可以精确求解薛定谔方程,获得振动波函数和能量。
图 2是 MgF 分子的电子态基态的 Morse 势函数图。
采用 Morse 势近似可得出当e R =R 时势能取得最小值,以及在核间距取值较大时的解离能e D ,这个和实际势能函数较为类似,但是在 R = 0 处,U 趋向于无穷大,这个在物理上是不成立的,而对于核间距值为零的势能函数是没有实际意义的。
图 2 MgF 电子态基态的 Morse 势函数要求解出上述的径向方程,可以将它改写成'ψ'+0=∂Ω∂ψ,其中''ψ表示为ψ的3阶导数,2B(R)ψ21 =ψ)Ω(R,,U)-2(E =B(R)。
若将 R 看成是类似于时间的变量,那么 ψ(R)和 ψ)Ω(R,就可以看成是广义坐标和势函数。
因此,可以将 (R) 'ψ' (R) ψ' 、分别看成是广义速度和广义加速度。
从而我们可以将方程 0 = ψΩ + 'ψ' ∂∂直接理解为某粒子在复空间中运动的牛顿方程。
假定 Lagrange 方程 B(R)ψ21 - ψ'2 1 = Ω - T =R), ψ'L(ψ( 22是关于'ψ的二次函数,函数 Ω、 T 分别等于 B(R)ψ21 ψ'2 122、,这样将R), ψ'L(ψ( 进行 Legendre 变换,从而可以得:),',('),,(R L R H ψψϕψϕψ-=(8)0)],',('['=-∂∂R L ψψϕψψ(9) 同时公式(9)可以得到:'''ψψϕ=∂∂=L (10) 将上述的公式ϕ代入到公式(8)当中,从而可以得到其哈密顿方程:22)(2121),,(ψϕϕψR B R H +=(11) 那么可以得到与(7)式相等价的哈密顿正则方程为:⎪⎩⎪⎨⎧=∂∂=-=ϕϕψψϕH R B ')('(12)根据哈密顿正则方程的空间演化可知,只需(12)式在1R 处的值,就可解得在2R 处的值,它们之间的演化关系为:⎥⎦⎤⎢⎣⎡=⎥⎦⎤⎢⎣⎡)()()()(212121R R g R R R R H ψϕψϕ(13)其中,)d 01)(0exp(1221R R B g R R R R H ⎰⎥⎦⎤⎢⎣⎡-= 可以看出(12)式具有 Symplectic 结构,那么可以考虑用 Symplectic 变换 对(13)式薛定谔方程进行求数值解。