系统发育树构建构建步骤
- 格式:ppt
- 大小:282.00 KB
- 文档页数:43


1、找模式菌株
Blast后,在LPSN内先找到属,再找到种,点击序列号,fasta后,复制文档至记事本,备注名称为(储藏所编号,序列号)
2、比对
打开GENEDOC,file-import-下载的序列,project-edit sequences list 删除不需要的序列,edit-pairwise alignment-align,edit-clear gap columns,+—号,人工比对(左键添加gaps,右键删除gaps),掐头去尾(edit-select columns-选择需要删除的末尾列-edit-delete all data)-file-export
输出的序列----最上面一行输入序列长度,然后将文档中的>和点号替换掉,除blast登录号外,其他的删除。
3、建树
打开TREECONW,找到treeconw exe打开,distantce estimation –start distantce estimation-找到文档,选择所有文件-打开目标文件-select all-taken into account,yes-ok-boots samples-1000,
后面选择YES,OK。最后draw phylogenetic tree,点file下的空白。标尺0.1,统计学50%。File-copy-任一word文档
4、修改名字
种名(斜体),保藏号,模式菌株加上标T,括号blast序列号

如何建树
step 1. 将16S rDNA序列在NCBI上进行BLAST比对(/BLAST/)
BLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990 [62];1997[63])。国际著名生物信息中心都提供基于Web的BLAST服务器。BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为内核向两端延伸,以找出尽可能长的相似序列片段。 首先登录到提供BLAST服务的常用网站,比如国内的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。这些网站提供的BLAST服务在界面上差不多,但所用的程序有所差异。它们都有一个大的文本框,用于粘贴需要搜索的序列。把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。如果是DNA序列,一般选择BLASTN搜索DNA数据库。
这里以NCBI为例。登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。
BLASTN结果如何分析(参数意义):
例如:
>gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete
sequence
Score = 2020 bits (1019), Expect = 0.0
Identities = 1382/1497 (92%), Gaps = 8/1497 (0%)
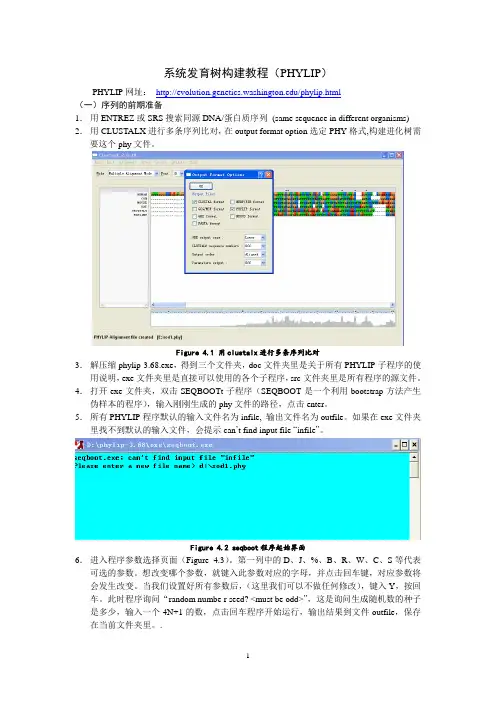
1 系统发育树构建教程(PHYLIP)
PHYLIP网址: /phylip.html
(一)序列的前期准备
1. 用ENTREZ或SRS搜索同源DNA/蛋白质序列 (same sequence in different organisms)
2. 用CLUSTALX进行多条序列比对,在output format option选定PHY格式,构建进化树需要这个phy文件。
Figure 4.1 用clustalx进行多条序列比对
3. 解压缩phylip-3.68.exe,得到三个文件夹,doc文件夹里是关于所有PHYLIP子程序的使用说明,exe文件夹里是直接可以使用的各个子程序,src文件夹里是所有程序的源文件。
4. 打开exe文件夹,双击SEQBOOTt子程序(SEQBOOT是一个利用bootstrap方法产生伪样本的程序),输入刚刚生成的phy文件的路径,点击enter。
5. 所有PHYLIP程序默认的输入文件名为infile, 输出文件名为outfile。如果在exe文件夹里找不到默认的输入文件,会提示can’t find input file “infile”。
Figure 4.2 seqboot程序起始界面
6. 进入程序参数选择页面(Figure 4.3)。第一列中的D、J、%、B、R、W、C、S等代表可选的参数。想改变哪个参数,就键入此参数对应的字母,并点击回车键,对应参数将会发生改变。当我们设置好所有参数后,(这里我们可以不做任何修改),键入Y,按回车。此时程序询问“random numbe r seed? ”,这是询问生成随机数的种子是多少,输入一个4N+1的数,点击回车程序开始运行,输出结果到文件outfile,保存在当前文件夹里。. 2
Figure 4.3 seqboot程序参数选择页面
主要参数解释:
D: 数据类型,有Molecular sequence、discrete morphology、restriction sites和gene frequencies4个选项。

upgma系统发育构建原理
UPGMA(Unweighted Pair Group Method with Arithmetic Mean)是一种常用的系统发育构建原理,它通过计算物种之间的遗传距离来构建进化树。UPGMA方法在遗传距离不准确或存在误差的情况下,可以得到一棵近似的进化树。
UPGMA方法的基本思想是将物种按照遗传距离的大小进行聚类,每次将距离最近的两个物种合并成一个新的聚类,直到所有物种都聚类为一棵进化树为止。这种方法假设进化速率是恒定的,并且所有物种是等距离分布的。
我们需要计算物种之间的遗传距离。遗传距离是通过测量不同物种之间的遗传差异来计算的,可以使用分子标记数据(如DNA序列或蛋白质序列)来进行计算。然后,根据遗传距离构建一个距离矩阵,矩阵中的每个元素表示两个物种之间的遗传距离。
接下来,我们需要选择距离最近的两个物种进行合并。合并的原则是选择距离最近的两个物种,计算它们的平均距离,并将它们合并成一个新的聚类。新的聚类将代替原来的两个物种,并更新距离矩阵中的相应值。
然后,重复上述步骤,直到所有物种都被聚类为止。每次合并两个物种后,都会更新距离矩阵中的值。最终,我们将得到一棵进化树,树的叶子节点代表每个物种,内部节点代表聚类。
UPGMA方法的优点是简单易用,特别适用于物种较少且进化速率基本恒定的情况。然而,UPGMA方法对遗传距离的准确性要求较高,如果遗传距离存在误差或不准确,可能会导致构建的进化树出现偏差。
UPGMA方法是一种常用的系统发育构建原理,通过计算物种之间的遗传距离来构建进化树。它的基本思想是将距离最近的两个物种合并成一个新的聚类,直到所有物种都聚类为一棵进化树。UPGMA方法简单易用,适用于物种较少且进化速率恒定的情况。然而,它对遗传距离的准确性要求较高,可能会受到误差的影响。