转录组数据分析解读及实例操作-1
- 格式:pdf
- 大小:4.07 MB
- 文档页数:49


转录组测序
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。转录组研究是基因功能及结构研究的基
础和出发点,通过新一代高通量测序,能够全面快速地获得某一物种特定组织或
器官在某一状态下的几乎所有转录本序列信息。
高通量转录组测序技术,无需了解物种基因信息,能够对任意物种进行转录
组分析,并接测定每个转录本的片段序列,精确到单核苷酸的分辨率;动态达6个数量级,能够同时鉴定和定量稀有转录本和正常转录本,以及检测基因家族中
相似基因和可变剪接造成的不同转录本的表达;被广泛的应用于转录本结构研究
(基因边界鉴定、可变剪切研究等),转录本变异研究(如基因融合、编码区SNP
研究),非编码区域功能研究(Non-coding RNA研究、microRNA前体研究等),
基因表达水平研究以及全新转录本发现。
转录组测序的优势 1 单次转录组测序实验即可提供全面的转录组信息
2 转录组测序无需预先知道任何序列信息
3 转录组测序提高了动态检测范围和灵敏度 4 转录组测序可提供转录本中序列变异信息
技术流程:
数据分析:
有参考基因组的转录组分析
1、测序数据质量评估
我们将测序得到的所有序列与目前常见物种进行大规模随机blast,检测可能的样品污染。 同时,我们对测序得到的序列进行深度和覆盖度的计算以评价
序列质量。
2、Reads比对到基因组 我们将测序结果与参考基因组进行比对, 比对的reads 中mismatch 的数
目可以根据客户需要进行分析, 并挑选出unique map的所有reads用于后续
peak的分析。同时进行reads的基因定位,用于后续分析。
3、Reads在基因组上的分布
4、新转录本的寻找
在每个样本中,都有一部分mapping不上已注释基因, 但是能mapping
上基因组上的reads,通过提取这些reads比对在基因组上的序列,利用同源预测
或者denevo预测,从而预测一些新的转录本。
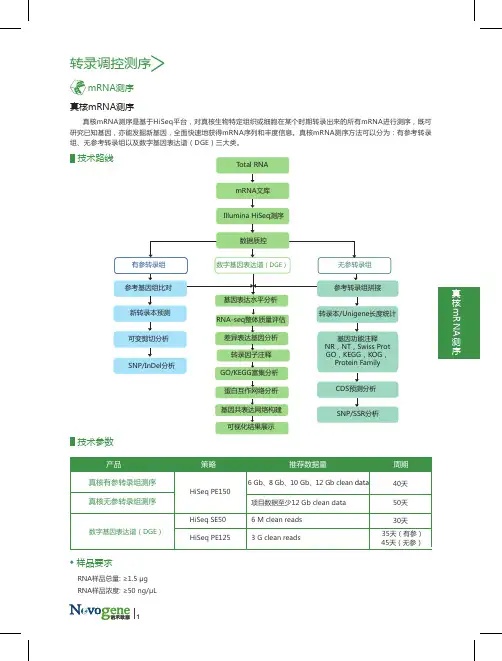
转录调控测序
mRNA测序
真核mRNA测序是基于HiSeq平台,对真核生物特定组织或细胞在某个时期转录出来的所有mRNA进行测序,既可研究已知基因,亦能发掘新基因,全面快速地获得mRNA序列和丰度信息。真核mRNA测序方法可以分为:有参考转录组、无参考转录组以及数字基因表达谱(DGE)三大类。真核mRNA测序
技术路线Total RNA
mRNA文库
Illumina HiSeq测序
参考基因组比对
新转录本预测
可变剪切分析
SNP/InDel分析参考转录组拼接
CDS预测分析
SNP/SSR分析基因功能注释NR,NT,Swiss ProtGO,KEGG,KOG,Protein Family转录本/Unigene长度统计基因表达水平分析
差异表达基因分析
GO/KEGG富集分析
基因共表达网络构建可视化结果展示数字基因表达谱(DGE)有参转录组无参转录组数据质控
真核mRNA
测序
技术参数
RNA样品总量: ≥1.5 μgRNA样品浓度: ≥50 ng/μL样品要求产品 策略 推荐数据量 周期真核有参转录组测序真核无参转录组测序HiSeq PE15040天50天 6 Gb、8 Gb、10 Gb、12 Gb clean data
30天6 M clean reads3 G clean reads35天(有参)项目数据至少12 Gb clean data
数字基因表达谱(DGE)HiSeq SE50HiSeq PE125 45天(无参)RNA-seq整体质量评估
转录因子注释
蛋白互作网络分析
1
诺禾携手华中农业大学,利用转录组测序和信息分析技术,研究了TDCPP处理组和对照组差异基因表达,并对差异表达基因进行KEGG通路分析,发现核糖体基因通路显著富集, 同时伴随胞浆和粗面内质网上核糖体数量减少体积增大。这些探索表明四膜虫可以作为TDCPP反应的生物指标,为后续研究TDCPP作用其他生物的毒理机制提供了新视角。诺禾致源携手中国农业科学院作物科学研究所,利用转录组测序技术,对杂交亲本、新合成异源六倍体小麦的幼苗、穗和种子进行了mRNA和smallRNA测序及信息分析,发现新合成异源六倍体小麦绝大部分基因表现为12类基因表达模式,包括加性表达,少部分的基因表现为非加性,基因的非加性表现出非常强的发育时期特异性,与生长势密切相关;miRNA的丰度随着倍性的增加逐渐下降,新合成异源六倍体小麦中非加性表达的 miRNA也同样表现出亲本显性表达,miRNA的表达敏感性与生长势和适应性密切相关。该研究揭示了不同倍性非对等杂种优势的分子基础。

转录组学验证模型-概述说明以及解释
1.引言
1.1 概述
转录组学验证模型是一种被广泛应用于生物学研究中的实验方法。随着基因组学和转录组学领域的不断发展,研究人员需要一种可靠的手段来验证和分析大量得到的转录组数据。转录组学验证模型应运而生,为研究人员提供了一种简便有效的工具。
在转录组学中,转录组是指一个生物体在某个时刻、某个条件下所有的转录本(mRNA)的总和。转录组学的研究旨在揭示基因的表达模式和调控机制,帮助我们理解生物体在不同环境中的生理和病理过程。
转录组学验证模型的设计与建立是实现这一目标的关键步骤之一。通过建立合适的验证模型,研究人员可以对转录组数据进行筛选、分析和解释,从而得到更加准确和可靠的实验结果。这些验证模型通常基于统计学和机器学习等方法,能够帮助研究人员挖掘和发现转录组中的重要信息。
转录组学验证模型的应用与实验结果也是该领域的研究重点之一。通过将验证模型应用于实际实验数据中,研究人员可以评估模型的性能和效果,并进一步验证和解释相关生物学问题。这些实验结果将为我们提供更深入的生物学认识,为相关领域的进一步研究和应用提供有力支持。
综上所述,转录组学验证模型在生物学研究中具有重要的意义。它不仅可以帮助我们更好地理解和解释转录组数据,还可以为我们提供有力的实验工具和方法。随着技术的不断进步和思想的不断创新,相信转录组学验证模型将会在生物学研究中发挥更加重要和广泛的作用。
1.2文章结构
1.2 文章结构
本文将按照以下结构展开讨论转录组学验证模型的设计与应用。首先,我们将在第2.1节介绍转录组学的基本概念,以帮助读者全面了解相关背景知识。接下来,在第2.2节中,我们将详细阐述转录组学验证模型的设计与建立过程,包括实验设计、样本采集、转录组测序和数据分析等关键步骤。值得注意的是,我们将重点介绍转录组学验证模型设计中的理论基础和方法策略,以及如何应对可能出现的挑战和问题。
在第2.3节,我们将展示转录组学验证模型在实际应用中的成果和实验结果。通过具体的案例和数据分析,我们将探讨转录组学验证模型在疾病诊断、药物研发、基因功能研究等领域的应用潜力,并展示其在解决当前研究问题中的有效性和可行性。此外,我们还将对转录组学验证模型的局限性和未来发展方向进行深入讨论,并提出相关建议和展望。

宏转录组测序流程-概述说明以及解释
1.引言
1.1 概述
概述
宏转录组测序是一种高通量的技术,可以同时检测样本中的所有转录本,从而了解基因表达的全貌。它在生物学研究、疾病诊断和药物开发等领域具有重要的应用价值。
随着测序技术的不断发展和成本的降低,宏转录组测序已经成为研究基因表达的重要方法之一。相比传统的基因表达分析方法,宏转录组测序具有高通量、高灵敏度、高准确性的特点,可以同时分析成千上万个基因的表达情况。
通过宏转录组测序,我们可以全面了解一个生物样本中的转录组信息,包括哪些基因被表达、不同基因的表达水平以及表达的调控网络等。通过对不同样本的转录组数据进行比较分析,我们可以发现与某种生理状态或疾病相关的基因,找出潜在的治疗靶点或疾病生物标志物。
宏转录组测序的流程包括样本准备、RNA提取、cDNA合成、文库构建、测序和数据分析等多个步骤。其中,样本准备和RNA提取是关键的步骤,不同的样本来源和实验目的需要不同的处理方法。cDNA合成和文库构建是将RNA转录本转化为测序可读的DNA片段的关键步骤,文库的质量将直接影响后续测序的准确性和可靠性。测序和数据分析是宏转录组测序的关键环节,选择适当的测序平台和对测序数据进行准确的比对和差异表达分析是确保数据质量和研究结果可靠性的重要步骤。
通过宏转录组测序,我们可以更全面地了解基因的表达调控网络,在生物学研究和医学诊断中具有广阔的应用前景。然而,宏转录组测序仍然面临着一些挑战,如数据分析的复杂性、样本的准备和RNA提取的高标准要求等。随着技术的进一步发展和改进,相信宏转录组测序将会在基因表达研究中发挥越来越重要的作用。
1.2 文章结构
文章结构是指文章的组织架构和章节安排。一个良好的文章结构可以帮助读者更好地理解文章的内容,并使文章逻辑清晰、条理分明。本文将围绕宏转录组测序流程展开,分为引言、正文和结论三个部分。
引言部分主要包括概述、文章结构和目的三个方面。在概述中,我们将简要介绍什么是宏转录组测序以及其在生物科研领域中的重要性。接下来,我们将详细介绍本文的组织结构,以帮助读者更好地理解全文。最后,我们将明确本文的目的,即介绍宏转录组测序流程和其在基因组学研究中的应用。