DNAsp5计算序列之间的Ka
- 格式:pdf
- 大小:514.75 KB
- 文档页数:5
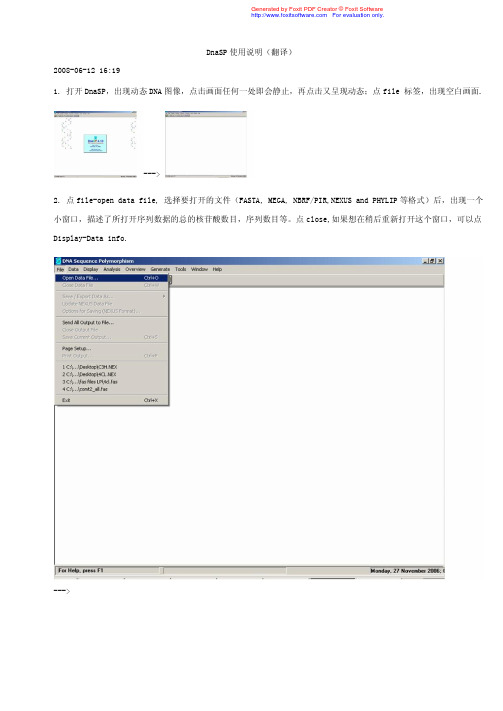
DnaSP使用说明(翻译)2008-06-12 16:191. 打开DnaSP,出现动态DNA图像,点击画面任何一处即会静止,再点击又呈现动态;点file 标签,出现空白画面.--->2. 点file-open data file, 选择要打开的文件(FASTA, MEGA, NBRF/PIR,NEXUS and PHYLIP等格式)后,出现一个小窗口,描述了所打开序列数据的总的核苷酸数目,序列数目等。
点close,如果想在稍后重新打开这个窗口,可以点Display-Data info.--->3. Display-view data,查看对比序列.4. 计算核苷酸多态性:点Analysis-DNA polymorphism, 出现一个选项卡供选择要分析序列的全长或某一区段(Region to Analyze)和运算法则(options)等;如果想知道在整个序列的哪一段多态性较高,可以在sliding window点中compute进行选择。
如果只打开了一个序列,则会出现要求选择多个序列进行分析的提示框.-5. 中性检验点击Analysis-Tajima's test该检验的目的是鉴定目标DNA序列在进化过程中是否遵循中性进化模型。
大多数由自发突变造成的分子差异不会影响到个体适宜度,推论指出基因进化根本上是通过遗传漂变来实现的。
Tajima's D 统计将序列分离位点数和两两序列差异平均数的估计进行对比。
在固定规模的试验群体中,如果这两个估计值相等,则只存在遗传漂变;反之,则是存在另一种选择方式影响了群体基因序列。
Tajima's D 统计值为正值时说明序列进化方式为平衡选择,且有一些单倍型分化;负值时为负向选择。
(Tajima Test(Tajima’s D)是由日本学者Fumio Tajima在1989年提出的。
该检验的目的是鉴定目标DNA序列在进化过程中是否遵循中性进化模型。

dnasp的原理及应用1. dnasp的介绍dnasp是一种用于分析DNA序列数据的计算工具,它可以帮助研究人员从DNA序列中获得有关遗传变异、种群结构、种群历史、选择压力等信息。
这些信息对于进化生物学、系统学和种群遗传学等领域的研究非常重要。
2. dnasp的原理dnasp主要基于DNA序列的比较和统计分析来推断分子进化、种群历史和遗传变异。
它使用了多种统计方法和模型,包括核苷酸多样性、基因多样性、分子变异网络等。
dnasp的基本原理如下:•DNA序列的比较:dnasp根据DNA序列的同源性和差异性进行分析。
它可以比较不同个体、不同种群和不同物种之间的DNA序列,从而揭示它们之间的遗传关系,并推断它们的进化历史。
•统计分析:dnasp使用统计方法来计算DNA序列的多样性和变异程度。
通过统计分析,它可以确定DNA序列的基因型频率、等位基因频率和基因型分布,从而揭示遗传变异的程度和种群结构。
•模型推断:dnasp使用进化模型来推断DNA序列的进化过程和种群历史。
它可以根据DNA序列的突变模式和频率,推断突变率、种群大小、遗传漂变等参数,从而研究不同进化过程和选择压力。
3. dnasp的应用dnasp在生物学研究中有广泛的应用。
以下是dnasp的几个主要应用领域:3.1 种群遗传学dnasp可以用于研究自然种群和人口的遗传变异和种群结构。
它可以分析DNA序列的多样性和变异,从而揭示种群的遗传多样性、基因流动和分化程度。
这对于保护濒危物种、研究种群动态和推断迁移历史等方面非常重要。
3.2 进化生物学dnasp可以用于研究物种的起源和进化过程。
它可以推断DNA序列的进化树和进化距离,从而揭示物种之间的亲缘关系和进化模式。
这对于研究进化机制、物种形成和适应性进化等方面非常重要。
3.3 系统学dnasp可以用于研究不同物种和亚种的遗传差异和分类关系。
它可以比较DNA 序列的差异、分析基因组的结构和演化,从而确定不同物种和亚种之间的亲缘关系和分类地位。
利用DnaSP计算Ka/Ks
2012年09月29日⁄ Bioinformatics ⁄字号小中大⁄评论1 条⁄阅读2,037 次[点击加入在线收藏夹]前面已经分享了一篇文章介绍《使用DnaSP计算核苷酸多样性和单倍型多样性》,最近刚好在用DnaSP v5,在写一篇文章与大家分享一下利用DnaSP 计算Ka、Ks,即dN,dS。
第一步:打开主界面之后,在File里面读取比对好的基因核苷酸序列(DnaSP支持fatsa,phylip等多种格式)
第二步:设置编码区域与密码子。
在data这一栏里面,有一个Assign Coding Regions的选项,在这里设定一下编码区域的其实位置,如果提交的序列全是DNA序列,可以设置为从序列的起始到终止。
点击后提示是否需要设置其他编码区域,可以选择否。
另外还需要设置一下使用的密码子表,这一项需要在Assign Genetic Code这一栏来选择,这里面有目前所有的密码子表。
第三步:在Analysis里面有一个选项是Synonymous and NonSynomymous Substitutions,点击这项进行Ka,Ks(即dN,dS)分析。
第四步:执行完第三步后,会出现两个结果页面,其一是pair-wise的基因Ka,Ks值得列表,例如:
另一个是以文本的形式展现的,额外还有其他的信息。



(完整版)DNA碱基计算⽅法总结DNA碱基计算⽅法总结对于碱基计算的这类题⽬,主要是抓住⼏个基础关系.只要能掌握三个关系式,灵活运⽤这三个关系式,就可解决⼀般的碱基计算的题⽬.这三个关系式分别是:A=T,C=G,A1=T2,A2=T1,C1=G2,C2=G1(1)在双链中,两种不互补的碱基和与另外两种的和相等。
A+G T+C =A+CT+G=1(2)双链中不互补的碱基和与双链碱基总数之⽐等于50%A+GA+T+G+C =A+CA+T+G+C=12(3)⼀条链中不互补碱基的和之⽐等于另⼀条链中这种⽐值的倒数。
A1+G1 T1+C1= a,A2+G2T2+C2=1a(4)⼀条链中互补碱基的和之⽐等于另⼀条链中这种⽐值,也等于双链中的这种⽐值。
A1+T1 G1+C1=a,则 A2+T2G2+C2=a,A+TG+C=a典型例题:1.分析⼀个DNA分⼦时,发现30%的脱氧核苷酸含有腺嘌呤,由此可知该分⼦中⼀条链上鸟嘌呤的最⼤值可占此链碱基总数的()A.20%B.30%C.40%D.70%2.现有⼀待测核酸样品,经检测后,对碱基个数统计和计算得到下列结果:(A+T)/(G+C)=1(A+G)/(T+C)=1根据此结果,该样品()A⽆法确定是脱氧核糖核酸还是核糖核酸B可被确定为双链DNAC⽆法被确定是单链DNA还是双链DNAD可被确定为单链DNA3.某双链DNA分⼦中,G占碱基总数的38%,其中⼀条链的T1占该DNA分⼦全部碱基总数的5%,那么另⼀条链的T2占该DNA 分⼦全部碱基⽐例为( )A、5%B、7%C、24%D、38%4.已知1个DNA分⼦中有4000个碱基对,其中胞嘧啶有2200个,这个DNA中应含有的脱氧核苷酸的数⽬和腺嘌呤的数⽬分别是:A、4000个和900个B、4000个和1800个C、8000个和1800个D、8000个和3600个5.⼀段多核苷酸链中的碱基组成为20%的A,20%的C,30%的G,30%的T。
细胞DNA序列分析中的统计方法随着科技的不断发展,人类对基因的认识越来越深入,而这种认识的基础就是对DNA序列的解析。
在DNA序列的分析中,统计学方法发挥了非常重要的作用,其在基因组学、遗传学等领域都有着广泛的应用。
本文将就细胞DNA序列分析中常用的几种统计方法进行介绍。
一、N元模型N元模型是一种常用的概率模型,在DNA序列分析中也有着广泛的应用。
在这种模型中,DNA序列中的每个碱基都被看成是一个离散的符号,N个相邻的碱基组成一组,被称为N元组。
这样就建立了一个由离散符号组成的序列,可以应用数学中的概率思想进行分析。
在N元模型中,每个N元组出现的概率可以通过计数来估算。
假设有一个长度为L的DNA序列S,其中包含k个N元组T1,T2,…,Tk,那么在该序列中T1出现的概率就是k1/L,T2出现的概率就是k2/L,以此类推。
这样计算出的概率可以用于后续的分析过程中,比如通过N元模型计算一个序列的信息熵,可以衡量序列的复杂程度和随机性。
二、k-mer计数在DNA序列中,k-mer是一种连续的k个碱基序列,也被称为k个字母的单词。
对于给定的k值,k-mer计数就是对一个DNA序列中所有长度为k的连续子序列进行计数。
这种方法可以用于对序列的特征进行描述,比如通过计算k-mer在序列中的出现频率来判断两个序列之间的相似度。
在实际应用中,k-mer计数可以通过哈希算法来加速计算。
哈希算法是一种常用的快速查找技术,将k-mer转换成一个哈希值,然后将哈希值作为索引进行存储。
这种方法可以大大加快计数过程,提高计算效率。
三、PWM模型PWM(Position Weight Matrix)模型也是一种常用的DNA序列分析方法,它可以用来描述某一蛋白质与DNA结合时的特征。
在PWM模型中,同样采用N元组的概念,但其可以更直观地表示一个N元组中各个碱基之间的关系。
举例来说,假设一个N元组A在序列中出现的概率为p(A),其包含了k个碱基(假设k=4),则可以构建一个4×4的矩阵M,其中第i行第j列的元素表示碱基i在A的第j个位置上出现的概率,即M(i,j)=p(Aj=i)。
基于互模式熵的DNA序列相似性分析
DNA序列相似性分析是计算生物学领域中的一个重要研究方向,它可以帮助鉴定物种、研究进化关系、发现基因等。
互模式熵是一种用来评估序列相似性的方法,同时也能够同
批判地评估假阳性和假阴性的概率。
互模式熵是一种信息量度,可以用来描述两个或多个序列之间的相似性。
互模式熵有
助于比较序列片段的异同,即找出它们之间的区别。
它可以通过计算序列中所有相同位置
上的碱基对应的频率得到。
具体而言,互模式熵的计算方法如下:
H(A,B)=−∑i=1npi log2pi
其中n为序列长度, pi表示序列A和B在位置i处出现的概率。
通过计算互模式熵,我们可以得到序列A和B之间的距离,将这个距离表示为一个矩阵。
该矩阵可以用来构建一个树状图,表示序列A和B之间的进化路径。
除了互模式熵,还有许多其他方法可以用来计算DNA序列的相似性。
例如,我们可以
使用序列比对的方法来找出两个序列之间的同源区域或配准点,或者利用进化模型来评估
序列之间的相似性。
然而,许多方法的缺陷在于它们只能针对数个序列进行比对。
同时,在进行序列比对时,我们通常会面临误判的问题。
互模式熵的优势在于它可以为大量序列提供可靠的比较
结果,并且同时考虑假阳性和假阴性的风险。
总之,互模式熵是一种简便的、可靠的方法来计算DNA序列之间的相似性。
它在计算
效率、准确性和稳定性方面都表现良好,已被广泛应用于许多DNA序列分析的研究中。
碱基替换率--知识介绍dN/dS碱基替换率在遗传学中,Ka/Ks或者dN/dS表示的是异意替换(Ka)和同意替换(Ks)之间的比例。
这个比例可以判断是否有选择压力作用于这个蛋白质编码基因。
不导致氨基酸改变的核苷酸变异我们称为同义突变,反之则称为非同义突变。
一般认为,同义突变不受自然选择,而非同义突变则受到自然选择作用。
在进化分析中,了解同义突变和非同义突变发生的速率是很有意义的。
常用的参数有以下几种:同义突变频率(Ks)、非同义突变频率(Ka)、非同义突变率与同义突变率的比值(Ka/Ks)。
如果Ka/Ks>1,则认为有正选择效应。
如果Ka/Ks=1,则认为存在中性选择。
如果Ka/Ks<1,则认为有纯化选择作用。
Ks = 同义突变SNP数/同义位点数,即同义突变率Ka = 非同义突变SNP数/非同义位点数,即非同义突变率同义突变SNP数= Σ同义SNP非同义突变SNP数= Σ非同义SNP同义位点数= Σ同义位点非同义位点数= Σ非同义位点uKa>>Ks或者Ka/Ks >> 1,基因受正选择(positive selection) uKa=Ks或者Ka/Ks =1,基因中性进化(neutral evolution)uKa<<<="" bdsfid="73" ks="" p="" selection)<="">检测序列的功能性(funcional or pseudo)筛选正在快速进化的基因(rapid evolution)Ks可以反映事件发生的时间(age)非同义替换率(氨基酸改变,dn)与同义替换率的(氨基酸不改变,ds)的比值(dn/ds)也经常被用于分化分析。
dn/ds的比值为1表示所研究的基因在中性选择(neutralselection)下进化,小于0. 25意味着纯化选择(purifying selection)下进化,当比值大于1时则被认为进行正向选择(positive selection)下的进化(Hurst et al, 2002; Swanson et al 2003)。
[转载]ka/ks的计算步骤
(2013-04-25 17:47:30)
转载▼
分类:生物技术
标签:
转载
原文地址:ka/ks的计算步骤作者:limits999
A、安装PAML、MEGA和DAMBE软件;
B、从MEGA软件中的File中导入需进行比对的序列文件,点击Align(要求核苷酸序列是三的倍数,没有终止密码子,核苷酸序列的第一位是密码子的第一位);
C、在新打开的窗口中的Alignment按钮中选择序列比对的方法(可选择Align by ClustalW(Codons)或者Align by Muscle(Codons));
D、在新窗口中的Data按钮中选择序列比对结果的输出方式(建议选择Fasta格式);
E、打开DAMBE中的File按钮导入序列比对文件(选择open standard sequence file);
F、在DAMBE中的File按钮把文件转化成所需要的格式进行输出(选择Save or Convert Sequence Format,建议选择PAML格式,虽然phylip格式和nexus格式也可被PAML有条件的识别,但要按照PAML的要求进行改造:a、序列名称需不超过10个字符,b、序列名称和序列之间需有2个以上的空格间隔,c、如果是层叠式数据,需要加上I进行注释,其余类别数据同理处理);
G、把转换后的数据文件和PAML子程序yn00.exe和它的控制文件yn00.ctl放到相同的工作文件夹中;
H、对yn00.ctl文件的输入、输出项根据自己的输入、输出文件名进行相应修改;
I、打开命令行,把当前目录调整到工作文件夹中,输入yn00;
J、打开输出文件,即可查看结果。
DNAsp5计算序列之间的Ka/Ks
熊荣川
中国科学院成都生物研究所
首先,打开DNAsp5,如下图。
导入比对好的序列
跳出的数据信息框
很多都是默认信息,可能和你的数据信息不相符合,需要针对具体的数据进行具体设置
在格式format中进行设置
例如,是否为单倍体“Haploid”,是常染色体“Autosome”还是线粒体“Mitochondrial”等等
设置好之后点击“OK”
设置密码子表。
程序默认的密码子是核基因密码子表,因此如果我们导入的数据来自线粒体
或是叶绿体的话需要重新设置
然后设置编码区域。
有时候我们的序列包括外显子和内含子,而我们计算的同义替换及非同义替换率是针对编码区域的,因此需要对计算区域进行预定义
设置编码区域和起始密码子位置之后,“OK”最后一步就是Ka、Ks的计算。
结果。