附录生化药品
- 格式:docx
- 大小:36.45 KB
- 文档页数:9

药品生产质量管理规范生化药品附录生化药品附录第一章范围第一条本附录所指生化药品就是指从动物的器官、组织、体液、分泌物中经前处理、提取、分离、纯化等制得的安全、有效、质量可控的药品。
主要包括:蛋白质、多肽、氨基酸及其衍生物、多糖、核苷酸及其衍生物、脂、酶及辅酶等(不包括生物制品附录所列产品)。
第二条本附录适用于原材料的前处理、提取、分离、纯化等原料(原液)及其制剂的制备与质量控制的全过程。
原材料的采集过程应符合国家相关规定,药品生产企业应监督控制其来源及质量。
第三条来源于人的组织、尿液的产品按照本附录执行。
第二章原则第四条应建立完善的质量管理体系,依据质量风险管理的原则,结合品种特点,明确从原材料采集至成品放行各阶段的质量管理责任,确保产品的安全有效、质量可控。
第五条生化药品具有以下特殊性,应对原材料的来源及质量、生产过程、中间产品的检验进行特殊控制:(一)生化药品的生产涉及器官、组织、体液、分泌物的提取、分离与纯化等过程,原材料本身具有不均一性。
(二)生化药品的质量控制通常采用生物分析技术,比理化测定具有更大的可变性。
(三)生产过程中的原材料与中间产品就是污染微生物生长的良好培养基,原材料中的病原微生物对产品质量与生产环境存在较大风险。
第三章人员第六条从事生化药品生产、质量保证、质量控制、采购及其她相关人员(包括清洁、维修人员)均应根据其生产的产品与所从事的生产操作定期进行相关法律法规、专业知识、卫生与微生物学基础知识及安全防护要求等方面的培训及考核,并纳入个人培训档案。
第七条生产管理负责人、质量管理负责人与质量受权人应具有相应的专业知识(如微生物学、生物学、免疫学、药学、生物制药、生物化学等),并能够在生产、质量管理中切实履行职责。
从事供应商审计的人员,应了解动物种属、饲养、屠宰、检疫、采集及其原材料贮存运输等方面的相关知识,并能够在供应商管理与审核过程中有效履行其职责。
第八条应对所生产品种的生物安全进行评估,根据评估结果,生产、维修、检验的操作人员、管理人员应采取必要的生物安全防护措施。

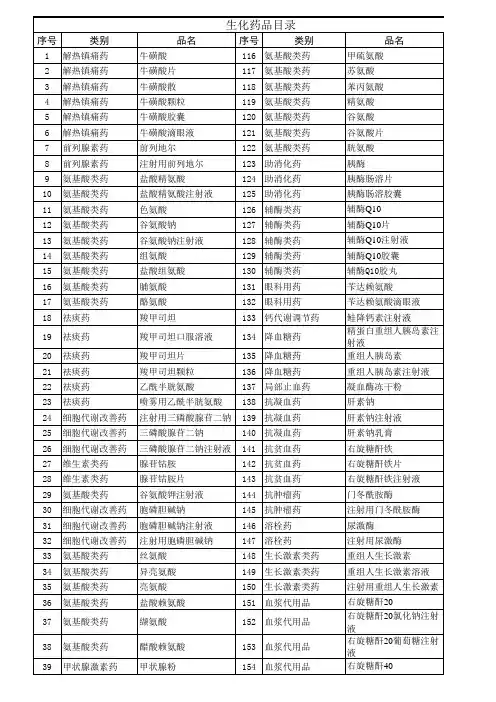
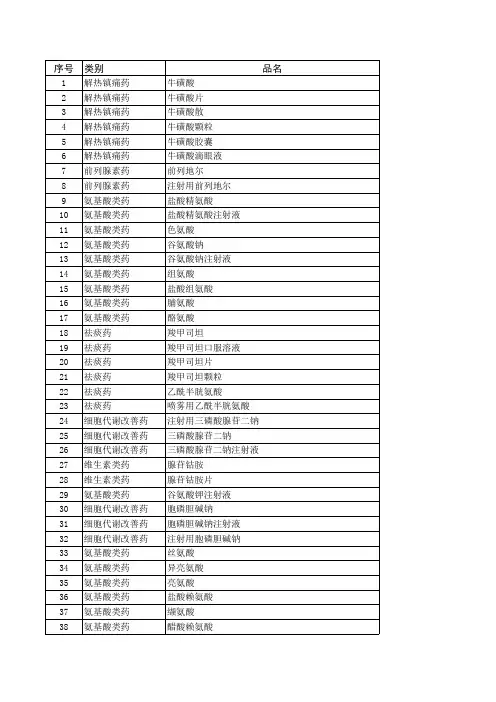
备注:本目录共包括2000药典二部所有品种,共1699条;包括部颁标准1-6册共1088条;新药转正标准1-药典2000二部,共1699条。
药品名称标准名称阿苯达唑中国药典2000年版二部阿苯达唑胶囊中国药典2000年版二部阿苯达唑颗粒中国药典2000年版二部阿苯达唑片中国药典2000年版二部艾司唑仑中国药典2000年版二部艾司唑仑片中国药典2000年版二部艾司唑仑注射液中国药典2000年版二部阿桔片中国药典2000年版二部阿米卡星中国药典2000年版二部阿莫西林中国药典2000年版二部阿莫西林胶囊中国药典2000年版二部阿莫西林克拉维酸钾片中国药典2000年版二部阿莫西林钠中国药典2000年版二部阿莫西林片中国药典2000年版二部氨苯蝶啶中国药典2000年版二部氨苯蝶啶片中国药典2000年版二部氨苯砜中国药典2000年版二部氨苯砜片中国药典2000年版二部氨苄西林中国药典2000年版二部氨苄西林钠中国药典2000年版二部氨茶碱中国药典2000年版二部氨茶碱缓释片中国药典2000年版二部氨茶碱片中国药典2000年版二部氨茶碱注射液中国药典2000年版二部氨酚待因片(I)中国药典2000年版二部氨酚待因片(II)中国药典2000年版二部氨甲环酸中国药典2000年版二部氨甲环酸胶囊中国药典2000年版二部氨甲环酸片中国药典2000年版二部氨甲环酸注射液中国药典2000年版二部氨鲁米特中国药典2000年版二部氨鲁米特片中国药典2000年版二部安乃近中国药典2000年版二部安乃近滴剂中国药典2000年版二部安乃近片中国药典2000年版二部安乃近注射液中国药典2000年版二部安钠咖注射液中国药典2000年版二部奥沙普秦中国药典2000年版二部奥沙西泮中国药典2000年版二部奥沙西泮片中国药典2000年版二部阿片中国药典2000年版二部阿片酊中国药典2000年版二部阿片粉中国药典2000年版二部阿普唑仑中国药典2000年版二部阿普唑仑片中国药典2000年版二部阿奇霉素中国药典2000年版二部阿奇霉素干混悬剂中国药典2000年版二部阿奇霉素胶囊中国药典2000年版二部阿奇霉素片中国药典2000年版二部。

⽣化药品⽣化药品所谓⽣化药品是指运⽤⽣物化学研究成果,由⽣物体中起重要⽣理⽣化作⽤的各种基本物质经过提取、分离、纯化等⼿段制造出的药物,或由上述这些已知药物加以结构改造或⼈⼯合成创造出的⾃然界所没有的新药物。
由于约定俗成,⽣化药物不包括抗⽣素(抗⽣素早已⾃成体系);也不包括⽤细菌疫苗制成的供预防、治疗和诊断特定传染病或其它有关疾病的⽣物制品;习惯上也不包括植物药中提取的⽣物碱。
⽣化类药物分为氨基酸类、酶类、核酸类、糖类、脂质类、多肽及蛋⽩质类六种。
药物的品种⾮常多,每类中常⽤药物均有近⼆⼗种。
据统计,中国⽣化制药⼯业年产值只有40多个亿,只占整个制药⾏业产值的4%。
克林霉素磷酸酯注射液属注射剂类抗菌素。
⽤于⾰兰阳性菌引起的下列各种感染性疾病如扁桃体炎、化脓性中⽿炎、⿐窦炎等。
急性⽀⽓管炎、慢性⽀⽓管炎急性发作、肺炎、肺脓肿和⽀⽓管扩张合并感染等。
⽪肤和软组织感染,泌尿系统感染:急性尿道炎、急性肾盂肾炎、前列腺炎等。
⾻髓炎、败⾎症、腹膜炎和⼝腔感染等。
亦可⽤于厌氧菌引起的各种感染性疾病,如脓胸、肺脓肿、厌氧菌性肺炎。
⽪肤和软组织感染、败⾎症。
强肝胶囊强肝胶囊的主要功能是清热利湿,补脾养⾎,益⽓解郁。
⽤于慢性肝炎,早期肝硬化,脂肪肝,中毒性肝炎等。
饭后⼝服,⼀次5粒,⼀⽇2次或遵医嘱,每服6⽇停⼀⽇,8周为⼀疗程,停⼀周再进⾏第⼆疗程。
慢性肝炎⼀般应⽤3-5个疗程肿节风胶囊本品具有祛风通络、活⾎化淤的功效,并有抗菌、消炎作⽤,临床⽤于治疗肺炎、扁桃体炎、胃肠炎、⼝腔炎及外伤感染等。
近年来⽤于治疗肿瘤有⼀定效果。
对胰腺癌、胃癌、直肠癌、肝癌、⾷道癌、⿐咽癌等癌症患者有改善直觉症状、增加⾷欲、延长⽣存期、减少并发症、减轻疼痛等作⽤,本品的不良反应少,适于长期服⽤,另外对胃溃疡有显著的治疗作⽤。
2、本品在内科⽤于胃溃疡、⼗⼆指肠溃疡、胃炎、肠炎;五官科⽤于治疗各类炎症及⽤于妇科炎症,均有显著疗效。
此外对甲沟炎、脓胞疮、病毒性疱疹及烧伤等外科感染均有较好的疗效。

生化药品附录第一章范围第一条本附录所指生化药品就是指从动物的器官、组织、体液、分泌物中经前处理、提取、分离、纯化等制得的安全、有效、质量可控的药品。
主要包括:蛋白质、多肽、氨基酸及其衍生物、多糖、核苷酸及其衍生物、脂、酶及辅酶等(不包括生物制品附录所列产品)。
第二条本附录适用于原材料的前处理、提取、分离、纯化等原料(原液)及其制剂的制备与质量控制的全过程。
原材料的采集过程应符合国家相关规定,药品生产企业应监督控制其来源及质量。
第三条来源于人的组织、尿液的产品按照本附录执行。
第二章原则第四条应建立完善的质量管理体系,依据质量风险管理的原则,结合品种特点,明确从原材料采集至成品放行各阶段的质量管理责任,确保产品的安全有效、质量可控。
第五条生化药品具有以下特殊性,应对原材料的来源及质量、生产过程、中间产品的检验进行特殊控制:(一)生化药品的生产涉及器官、组织、体液、分泌物的提取、分离与纯化等过程,原材料本身具有不均一性。
(二)生化药品的质量控制通常采用生物分析技术,比理化测定具有更大的可变性。
(三)生产过程中的原材料与中间产品就是污染微生物生长的良好培养基,原材料中的病原微生物对产品质量与生产环境存在较大风险。
第三章人员第六条从事生化药品生产、质量保证、质量控制、采购及其她相关人员(包括清洁、维修人员)均应根据其生产的产品与所从事的生产操作定期进行相关法律法规、专业知识、卫生与微生物学基础知识及安全防护要求等方面的培训及考核,并纳入个人培训档案。
第七条生产管理负责人、质量管理负责人与质量受权人应具有相应的专业知识(如微生物学、生物学、免疫学、药学、生物制药、生物化学等),并能够在生产、质量管理中切实履行职责。
从事供应商审计的人员,应了解动物种属、饲养、屠宰、检疫、采集及其原材料贮存运输等方面的相关知识,并能够在供应商管理与审核过程中有效履行其职责。
第八条应对所生产品种的生物安全进行评估,根据评估结果,生产、维修、检验的操作人员、管理人员应采取必要的生物安全防护措施。

我国GMP附录《生化药品》简析贾晓艳(奥星制药设备(石家庄)有限公司,河北石家庄050000)摘要:从范围、原则、人员、厂房与设备、病毒去除/灭活及验证、供应链管理、生产管理、质量管理、术语9个方面,对我国GMP附录《生化药品》进行了解读,以期为国内相关企业提供必要的帮助。
关键词:GMP;生化药品;附录;CFDA1概述2017年3月16日,国家食品药品监督管理总局(CFDA)发布了《药品生产质量管理规范(2010年修订)》附录《生化药品》的公告(2017年第29号),说明了《生化药品》附录(以下简称为该附录)为《药品生产质量管理规范》(2010年修订)的配套文件,并且自2017年9月1日起施行。
该附录曾于2016年10月19日发布了第一次征求意见稿,并在2016年11月12日征求意见结束。
该附录内容共44条,其主体部分共分为八章(有43条),第九章为术语解释。
2章节解析2.1第一章:范围本章对生化药品的定义、范围、生化药品的产品类型与工艺的范围、原材料的采集过程和药品生产企业的责任等内容提出了要求。
本章还对来源于人的组织、尿液的生化药品进行了说明,需要注意的是,血液制品不在该附录范围内。
典型的生化药品包括了肝素钠、辅酶Q10、氨基酸及其衍生物、胰酶、玻璃酸钠(鸡冠提取物)、蛋白质、多肽等。
典型的生化药品生产方法包含了提取、酶解、盐析、吸附、有机溶剂分级沉淀等。
需要注意的是,生化药品和生物制品是有很大区别的。
生物制品是用病原微生物(细菌、病毒、立克次氏体)、病原微生物的代谢产物(毒素)以及动物和人的血浆等制成的制品,可用于预防、治疗和诊断疾病。
其中,用于防治传染病的生物制品可分为人工自动免疫制品(如疫苗和类毒素等)和人工被动免疫制品(如丙种球蛋白、白喉抗毒素、破伤风抗毒素等)。
2.2第二章:原则本章对质量管理体系的管理范围和管控阶段提出了要求,并且要求用质量风险管理的方法来确保产品的安全有效、质量可控。

生化药品附录文稿归稿存档编号:[KKUY-KKIO69-OTM243-OLUI129-G00I-FDQS58-附件生化药品附录第一章范围第一条本附录所指生化药品是指从动物的器官、组织、体液、分泌物中经前处理、提取、分离、纯化等制得的安全、有效、质量可控的药品。
主要包括:蛋白质、多肽、氨基酸及其衍生物、多糖、核苷酸及其衍生物、脂、酶及辅酶等(不包括生物制品附录所列产品)。
第二条本附录适用于原材料的前处理、提取、分离、纯化等原料(原液)及其制剂的制备和质量控制的全过程。
原材料的采集过程应符合国家相关规定,药品生产企业应监督控制其来源及质量。
第三条来源于人的组织、尿液的产品按照本附录执行。
第二章原则第四条应建立完善的质量管理体系,依据质量风险管理的原则,结合品种特点,明确从原材料采集至成品放行各阶段的质量管理责任,确保产品的安全有效、质量可控。
第五条生化药品具有以下特殊性,应对原材料的来源及质量、生产过程、中间产品的检验进行特殊控制:(一)生化药品的生产涉及器官、组织、体液、分泌物的提取、分离和纯化等过程,原材料本身具有不均一性。
(二)生化药品的质量控制通常采用生物分析技术,比理化测定具有更大的可变性。
(三)生产过程中的原材料和中间产品是污染微生物生长的良好培养基,原材料中的病原微生物对产品质量和生产环境存在较大风险。
第三章人员第六条从事生化药品生产、质量保证、质量控制、采购及其他相关人员(包括清洁、维修人员)均应根据其生产的产品和所从事的生产操作定期进行相关法律法规、专业知识、卫生和微生物学基础知识及安全防护要求等方面的培训及考核,并纳入个人培训档案。
第七条生产管理负责人、质量管理负责人和质量受权人应具有相应的专业知识(如微生物学、生物学、免疫学、药学、生物制药、生物化学等),并能够在生产、质量管理中切实履行职责。
从事供应商审计的人员,应了解动物种属、饲养、屠宰、检疫、采集及其原材料贮存运输等方面的相关知识,并能够在供应商管理和审核过程中有效履行其职责。


其它药品标准卫生部颁药品标准(生化药品第一册)(38种)肝素钙注射液拼音名:Gansugai Zhusheye英文名:INJECTIO HEPARINI CALCII书页号:S1-15 标准编号:WS1-C3-0011-89 本品为肝素钙的灭菌水溶液。
其效价应为标示量的86.0~116.0%。
【性状】本品为无色或淡黄色的澄明液体。
【鉴别】照肝素钙项下的鉴别法试验,显相同的结果。
【检查】pH值应为5.0~8.0(中国药典1985年版二部附录33页)。
热原照肝素钙项下的方法检查,应符合规定。
其他应符合注射剂项下有关的各项规定(中国药典1985年版二部附录4 页)。
【效价测定】照肝素生物检定法测定(中国药典1985年版二部附录58页)。
【作用与用途】同肝素钙。
【用法与用量】皮下注射一次5,000~10,000单位一日一次。
【注意】用药期间应定时测定凝血时间。
有出血倾向及凝血机制障碍者禁用。
孕妇及产后妇女慎用。
【规格】(1) 1ml:5,000单位(2) 1ml:10,000单位(3) 2ml:10,000单位【贮藏】避光,在阴凉处保存。
有效期三年。
L-盐酸精氨酸拼音名:Yansuan Jing※ansuan英文名:L-ARGININI MONOHYDROCHLORIDUM书页号:S1-31 标准编号:WS1-C3-0022-89[C6H14N4O2·HCl=210.66] 本品为2-氨基-5-胍基戊酸盐酸盐。
按干燥品计算,含C6H14N4O2.HCl应为98.5~101.5%。
【性状】本品为白色结晶性粉末,水溶液显酸性反应。
本品在水中易溶,在乙醇中极微溶解。
比旋度取本品,精密称定,加盐酸溶液(9→100)制成每1ml中含30mg的溶液,依法测定(中国药典1985年版二部附录16页),按干燥品计算,比旋度应为+20.4°至+22.4°。
【鉴别】(1)取本品约2mg,加水2ml使溶解,加茚三酮约2mg,加热,溶液显蓝紫色。
化学药品质量控制按剂型分ⅠA片剂ⅠB注射剂ⅠC酊剂ⅠD栓剂ⅠE胶囊剂ⅠF软膏剂乳膏剂糊剂ⅠG眼用制剂ⅠH丸剂ⅠJ植入剂(增订)ⅠK糖浆剂ⅠL气雾剂粉雾剂喷雾剂ⅠM膜剂ⅠN颗粒剂ⅠO口服溶液剂口服混悬剂口服乳剂ⅠP散剂ⅠQ耳用制剂ⅠR鼻用制剂ⅠS洗剂冲洗剂灌肠剂ⅠT搽剂涂剂涂膜剂ⅠU凝胶剂ⅠV贴剂片剂系指药物与适宜的辅料混匀压制而成的圆片状或异形片状的固体制剂。
片剂以口服普通片为主,另有含片、舌下片、口腔贴片、咀嚼片、分散片、可溶片、泡腾片、阴道片、阴道泡腾片、缓释片、控释片与肠溶片等。
含片系指含于口腔中,药物缓慢溶解产生持久局部作用的片剂。
含片中的药物应是易溶性的,主要起局部消炎、杀菌、收敛、止痛或局部麻醉作用。
含片应进行释放度检查。
舌下片系指置于舌下能迅速溶化,药物经舌下黏膜吸收发挥全身作用的片剂。
舌下片中的药物与辅料应是易溶性的,主要适用于急症的治疗。
口腔贴片系指粘贴于口腔,经黏膜吸收后起局部或全身作用的片剂。
口腔贴片应进行溶出度或释放度检查。
咀嚼片系指于口腔中咀嚼或吮服使片剂溶化后吞服,在胃肠道中发挥作用或经胃肠道吸收发挥全身作用的片剂。
咀嚼片口感、外观均应良好,一般应选择甘露醇、山梨醇、蔗糖等水溶性辅料作填充剂和黏合剂。
咀嚼片的硬度应适宜。
分散片系指在水中能迅速崩解并均匀分散的片剂。
分散片中的药物应是难溶性的。
分散片可加水分散后口服,也可将分散片含于口中吮服或吞服。
分散片应进行溶出度检查。
可溶片系指临用前能溶解于水的非包衣片或薄膜包衣片剂。
可溶片应溶解于水中,溶液可呈轻微乳光。
可供外用、含漱等用。
泡腾片系指含有碳酸氢钠和有机酸,遇水可产生气体而呈泡腾状的片剂。
泡腾片中的药物应是易溶性的,加水产生气泡后应能溶解。
有机酸一般用枸橼酸、酒石酸、富马酸等。
阴道片与阴道泡腾片系指置于阴道内应用的片剂。
阴道片和阴道泡腾片的形状应易置于阴道内,可借助器具将阴道片送入阴道。
阴道片为普通片,在阴道内应易融化、崩解并释放药物,主要起局部消炎杀菌作用,也可给予性激素类药物。
资料二GMP生化药品附录(征求意见稿20160725)第一章范围第一条【定义】本附录所指生化药品是指从动物的器官、组织、体液、分泌物中经提取、分离、纯化等制得的安全、有效、质量可控的药品。
主要包括:蛋白质、多肽、氨基酸及其衍生物、多糖、核酸、核苷酸及其衍生物、脂、酶及辅酶等(不包括生物制品附录所列产品)。
来源于人的组织或体液的产品参照本附录执行(血液制品除外)。
第二条【范围】本附录适用于生化药品经粗加工后的器官、组织、体液、分泌物提取、分离、纯化等原液(原料)的制备过程,以及完成制剂的过程。
器官、组织、体液、分泌物的采集、切割、混合等加工过程应符合国家相关规定,药品生产企业应监督控制来源及质量。
生化药品的原料(原液)制备及制剂生产过程、质量控制应当符合本附录要求和国家相关规定。
第二章原则第三条【基本原则】企业应建立完善的质量管理体系,依据质量风险管理的原则,结合品种特点,明确从器官、组织、体液、分泌物采集至所生产药品各阶段的质量管理责任,确保产品的安全有效、质量可控。
第四条【质量控制原则】生化药品具有以下特殊性,应当对原材料的来源及质量、生产过程、中间产品的检验进行特殊控制:(一)生化药品的生产涉及从器官、组织、体液、分泌物中提取、分离和纯化等过程,原材料本身的不均一性使生产过程存在可变性。
因此应采用质量风险管理原则,制定原材料的质量控制措施。
(二)生化药品的质量控制通常涉及生物分析技术,比理化测定具有更大的可变性。
其质量控制更依赖于可靠的供应链管理、稳定的生产工艺和合理的中控措施。
(三)生产过程中的原材料和中间产品是污染微生物生长的良好培养基,原材料中的病毒对产品质量和生产环境存在较大风险。
因此在生产过程中应采取措施防止微生物滋生和病毒的扩散。
第三章人员第五条【基本要求】从事生化药品生产、质量保证、质量控制、采购及其他相关人员(包括清洁、维修人员)均应根据其生产的产品和所从事的生产操作定期进行相关法律法规、专业知识、卫生和微生物学基础知识及安全防护要求等方面的培训及考核,并纳入个人培训档案。
卫生部颁药品标准(生化药品)中华人民共和国卫生部药品标准(二部)第六册(生化药品第一分册)(生化药品第一分册)标准药品名称曾用名称页码三磷酸腺苷二钠…………………… 1 药品标准二部第六册(生化药品第一分册)三磷酸腺苷二钠注射液…………………… 3 药品标准二部第六册(生化药品第一分册)注射用三磷酸腺苷二钠…………………… 4 药品标准二部第六册(生化药品第一分册)口服胰糜酶口服清胃酶…………………… 5 药品标准二部第六册(生化药品第一分册)大豆磷脂大豆磷脂(口服乳剂)、…………………… 6 药品标准二部第六册(生化药品第一分册)大豆磷脂(注射用)、精制豆磷脂干酵母…………………… 8 药品标准二部第六册(生化药品第一分册)干酵母片…………………… 9 药品标准二部第六册(生化药品第一分册) L-门冬氨酸…………………… 10 药品标准二部第六册(生化药品第一分册)门冬氨酸钾镁口服溶液…………………… 12 药品标准二部第六册(生化药品第一分册)L-门冬酰胺酶 L-天门冬酰胺酶…………………… 13药品标准二部第六册(生化药品第一分册)注射用L-门冬酰胺酶注射用L-天门冬酰胺酶…………………… 16 药品标准二部第六册(生化药品第一分册)门冬酰胺天冬素…………………… 17 药品标准二部第六册(生化药品第一分册)门冬酰胺片天冬素片…………………… 19 药品标准二部第六册(生化药品第一分册)牛磺酸氨基己磺酸…………………… 20 药品标准二部第六册(生化药品第一分册)牛磺酸片氨基己磺酸片…………………… 21 药品标准二部第六册(生化药品第一分册)牛磺酸散氨基己磺酸散…………………… 22 药品标准二部第六册(生化药品第一分册)牛磺酸颗粒剂氨基己磺酸颗粒剂…………………… 23 药品标准二部第六册(生化药品第一分册)L-丙氨酸…………………… 24 药品标准二部第六册(生化药品第一分册)L-半胱氨酸盐酸盐盐酸半胱氨酸…………………… 26 药品标准二部第六册(生化药品第一分册)右旋糖酐20 小分子右旋糖酐…………………… 29 药品标准二部第六册(生化药品第一分册)右旋糖酐20葡萄糖注射液小分子右旋糖酐氯化钠注射液……………………30 药品标准二部第六册(生化药品第一分册)脉通氯化钠注射液右旋糖酐20氯化钠注射液小分子右旋糖酐葡萄糖注射液……………………31 药品标准二部第六册(生化药品第一分册)右旋糖酐铁…………………… 32 药品标准二部第六册(生化药品第一分册)右旋糖酐铁片…………………… 34 药品标准二部第六册(生化药品第一分册)右旋糖酐铁注射液…………………… 35 药品标准二部第六册(生化药品第一分册)多酶片…………………… 37 药品标准二部第六册(生化药品第一分册)肌苷…………………… 38 药品标准二部第六册(生化药品第一分册)肌苷口服液…………………… 40 药品标准二部第六册(生化药品第一分册)肌苷片…………………… 41 药品标准二部第六册(生化药品第一分册)肌苷注射液…………………… 42 药品标准二部第六册(生化药品第一分册)肌苷胶囊…………………… 43 药品标准二部第六册(生化药品第一分册) L-色氨酸…………………… 44 药品标准二部第六册(生化药品第一分册)利巴韦林三氮唑核苷、病毒唑…………………… 46 药品标准二部第六册(生化药品第一分册)利巴韦林片三氮唑核苷片、三氮唑核苷含片、…………………… 48 药品标准二部第六册(生化药品第一分册)病毒唑含片、三氮唑核苷口含片、药品标准二部第六册(生化药品第一分册)利巴韦林含片药品标准二部第六册(生化药品第一分册)利巴韦林注射液三氮唑核苷注射液、病毒唑针…………………… 49 药品标准二部第六册(生化药品第一分册)利巴韦林喷剂利巴韦林喷雾剂、三氮唑核苷喷雾…………………… 50 药品标准二部第六册(生化药品第一分册)剂、病毒唑喷雾剂药品标准二部第六册(生化药品第一分册)利巴韦林滴眼液三氮唑核苷滴眼液、三氮唑核苷眼…………………… 51 药品标准二部第六册(生化药品第一分册)药水、病毒唑眼药水药品标准二部第六册(生化药品第一分册)利巴韦林滴鼻液三氮唑核苷滴鼻液、三氮唑核苷滴…………………… 52 药品标准二部第六册(生化药品第一分册)鼻剂药品标准二部第六册(生化药品第一分册)肝素钙…………………… 53 药品标准二部第六册(生化药品第一分册)肝素钙注射液…………………… 54 药品标准二部第六册(生化药品第一分册) L-苏氨酸…………………… 55 药品标准二部第六册(生化药品第一分册)谷氨酸钠…………………… 57 药品标准二部第六册(生化药品第一分册)阿昔洛韦无环鸟苷…………………… 59 药品标准二部第六册(生化药品第一分册)阿昔洛韦软膏无环鸟苷软膏…………………… 61 药品标准二部第六册(生化药品第一分册)阿昔洛韦眼膏无环鸟苷眼膏…………………… 62 药品标准二部第六册(生化药品第一分册)阿昔洛韦滴眼液无环鸟苷滴眼液、无环鸟苷眼药水…………………… 63 药品标准二部第六册(生化药品第一分册)乳酶生…………………… 64 药品标准二部第六册(生化药品第一分册)乳酶生片…………………… 65 药品标准二部第六册(生化药品第一分册)乳酶菌素…………………… 66 药品标准二部第六册(生化药品第一分册)乳酶菌素片…………………… 67 药品标准二部第六册(生化药品第一分册)乳酶菌素散小儿乳酸菌素…………………… 68 药品标准二部第六册(生化药品第一分册)乳酶菌素颗粒剂乳酸菌素冲剂…………………… 69 药品标准二部第六册(生化药品第一分册)垂体后叶粉…………………… 70 药品标准二部第六册(生化药品第一分册)垂体后叶粉注射液…………………… 71 药品标准二部第六册(生化药品第一分册)L-组氨酸…………………… 72 药品标准二部第六册(生化药品第一分册)L-组氨酸盐酸盐盐酸组氨酸…………………… 74 药品标准二部第六册(生化药品第一分册)L-苯丙氨酸........................ 76 药品标准二部第六册(生化药品第一分册)复方氨基酸注射液(3AA) 3-氨基酸注射液、3H支链氨基酸注 (78)药品标准二部第六册(生化药品第一分册)射液、三合氨基酸注射液、肝脑清氨基酸注射液复方氨基酸注射液(9AA) 9-复合结晶氨基酸注射液、9合氨基 (79)药品标准二部第六册(生化药品第一分册)酸注射液、复合氨基酸-9R注射液、复方氨基酸-9R注射液、9种复方结晶氨基酸注射液、肾必安注射液、新肾必氨注射液复方氨基酸注射液(14AA) 14氨基酸注射液-823、14氨基酸注……………………81 药品标准二部第六册(生化药品第一分册)射液、氨复命14S注射液复方氨基酸注射液(15AA) 15合氨基酸注射液、15种氨基酸注……………………83 药品标准二部第六册(生化药品第一分册)射液、肝安注射液、14氨基酸注射液-800复方氨基酸注射液(17AA) 17-氨基酸注射液、复合氨基酸注射……………………85 药品标准二部第六册(生化药品第一分册)液17-725、17种结晶氨基酸注射液复方氨基酸注射液(18AA) 18-氨基酸注射液、18合氨基酸注射……………………88 药品标准二部第六册(生化药品第一分册)液、氨基酸注射液(18种)复方氨基酸注射液(18AA-?) 17种氨基酸注射液(凡命)……………………90药品标准二部第六册(生化药品第一分册)胃蛋白酶片…………………… 93 药品标准二部第六册(生化药品第一分册)胃蛋白酶颗粒剂胃蛋白酶冲剂…………………… 94 药品标准二部第六册(生化药品第一分册)胃膜素…………………… 95 药品标准二部第六册(生化药品第一分册)胆固醇…………………… 97 药品标准二部第六册(生化药品第一分册)胆酸钠…………………… 98 药品标准二部第六册(生化药品第一分册)胆酸钠片…………………… 99 药品标准二部第六册(生化药品第一分册)食母生片…………………… 100 药品标准二部第六册(生化药品第一分册)胞磷胆碱钠胞二磷胆碱钠…………………… 101 药品标准二部第六册(生化药品第一分册)胞磷胆碱钠注射液胞二磷胆碱钠注射液…………………… 103 药品标准二部第六册(生化药品第一分册)注射用胞磷胆碱钠注射用胞二磷胆碱钠…………………… 104 药品标准二部第六册(生化药品第一分册)莫海林降脂宁、净脉灵、改构肝素…………………… 105 药品标准二部第六册(生化药品第一分册)莫海林片降脂宁片、净脉灵片、改构肝素片…………………… 107 药品标准二部第六册(生化药品第一分册)氨糖美辛肠溶片复方消炎痛片…………………… 108 药品标准二部第六册(生化药品第一分册)胰酶肠溶胶囊…………………… 110 药品标准二部第六册(生化药品第一分册)胰激肽原酶胰激肽释放酶、血管舒缓素…………………… 111 药品标准二部第六册(生化药品第一分册)胰激肽原酶肠溶片胰激肽释放酶、胰激肽原酶片、血 (113)药品标准二部第六册(生化药品第一分册)管舒缓素片药品标准二部第六册(生化药品第一分册)注射用胰激肽原酶注射用胰激肽释放酶、注射用血管舒 (114)药品标准二部第六册(生化药品第一分册)缓素药品标准二部第六册(生化药品第一分册)L-胱氨酸…………………… 115 药品标准二部第六册(生化药品第一分册)脂肪乳注射液…………………… 117 药品标准二部第六册(生化药品第一分册)弹性酶…………………… 122 药品标准二部第六册(生化药品第一分册)弹性酶肠溶片弹性酶片…………………… 124 药品标准二部第六册(生化药品第一分册)羟己基淀粉20氯化钠注射液低分子羟乙基淀粉代血浆、低分子……………………125 药品标准二部第六册(生化药品第一分册)706代血浆、低分子羟乙基淀粉氯化钠注射液羟己基淀粉40氯化钠注射液羟乙基淀粉血浆、706代血浆、羟乙……………………126 药品标准二部第六册(生化药品第一分册)基淀粉氯化钠注射液L-脯氨酸…………………… 127 药品标准二部第六册(生化药品第一分册)L-蛋氨酸…………………… 129 药品标准二部第六册(生化药品第一分册)辅酶A …………………… 131 药品标准二部第六册(生化药品第一分册)注射用辅酶A …………………… 133 药品标准二部第六册(生化药品第一分册)辅酶Q10 …………………… 134 药品标准二部第六册(生化药品第一分册)辅酶Q11注射液…………………… 136 药品标准二部第六册(生化药品第一分册)辅酶Q10胶囊泛醌-10胶囊…………………… 137 药品标准二部第六册(生化药品第一分册)硫酸软骨素…………………… 138 药品标准二部第六册(生化药品第一分册)硫酸软骨素片…………………… 140 药品标准二部第六册(生化药品第一分册)溶菌酶…………………… 141 药品标准二部第六册(生化药品第一分册)溶菌酶含片溶菌酶口含片…………………… 143 药品标准二部第六册(生化药品第一分册)溶菌酶肠溶片…………………… 144 药品标准二部第六册(生化药品第一分册)羧甲司坦片(小儿用)小儿羧甲司坦片、小儿羧甲基半胱氨……………………145 药品标准二部第六册(生化药品第一分册)酸片羧甲淀粉钠溶液 405糖浆、卡慢舒溶液…………………… 146 药品标准二部第六册(生化药品第一分册)L-赖氨酸醋酸盐醋酸赖氨酸…………………… 148 药品标准二部第六册(生化药品第一分册)L-赖氨酸醋酸盐颗粒剂盐酸赖氨酸颗粒剂…………………… 150 药品标准二部第六册(生化药品第一分册)L-酪氨酸…………………… 151 药品标准二部第六册(生化药品第一分册)糜胰蛋白酶…………………… 153 药品标准二部第六册(生化药品第一分册)注射用糜胰蛋白酶…………………… 154 药品标准二部第六册(生化药品第一分册)附录升压素生物检定法…………………… 附录1 药品标准二部第六册(生化药品第一分册)灰分测定法…………………… 附录2 药品标准二部第六册(生化药品第一分册)多糖的分子量与分子量分布测定法…………………… 附录3 药品标准二部第六册(生化药品第一分册)乳酸菌制剂检验法…………………… 附录4 药品标准二部第六册(生化药品第一分册)羟乙基淀粉20(40)氯化钠注射液…………………… 附录7 药品标准二部第六册(生化药品第一分册)置换度测定法英文名称索引…………………… 索引1 药品标准二部第六册(生化药品第一分册)。
2010年版药典三部附录Ⅰ简介目录•1拼音•2附录Ⅰ A 注射剂•3附录Ⅰ B 栓剂•4附录Ⅰ C 眼用制剂•5附录Ⅰ D 外用制剂•6附录Ⅰ E 片剂•7附录Ⅰ F 胶囊剂•8附录Ⅰ G 软膏剂乳膏剂•9附录Ⅰ H 喷雾剂•10附录Ⅰ J 颗粒剂•11附录Ⅰ K 散剂•12附录Ⅰ L 鼻用制剂•13附录Ⅰ M 凝胶剂1拼音2010 nián bǎn yào diǎn sān bù fù lù Ⅰ《中华人民共和国药典》(2010年版)三部附录Ⅰ制剂通则本通则适用于治疗用生物制品,包括血液制品、免疫血清、细胞因子、单克隆抗体、免疫调节剂、微生态制剂等。
预防用生物制品,按品种项下的要求进行。
2附录Ⅰ A 注射剂注射剂系指以生物制品原液为原料药物,加入适宜稳定剂或其他辅料等制成的可供注入体内的无菌溶液、乳液、混悬液及临用前用无菌溶剂复溶为溶液、混悬液的无菌冻干制剂。
注射剂可分注射液、注射用无菌粉末。
注射液包括溶液型、乳液型或混悬型注射液。
可用于皮下注射、皮内注射、肌内注射、静脉注射和静脉滴注。
其中,供静脉滴注用的大体积(除另有规定外,一般不小于50ml)注射液也称静脉输液。
注射用无菌粉末系指供临用前以适宜的无菌溶液配制成澄明溶液或均匀混悬液的无菌固体制剂。
可用适宜的注射用溶剂配制后注射,也可用静脉输液配制后静脉滴注。
以冷冻干燥法制备的无菌粉末,称为注射用冻干制剂。
注射剂在生产和贮藏期间应符合下列有关规定。
(1)所用生物制品原液、半成品和成品的生产及质量控制应符合相关品种要求。
(2)注射剂所用的原辅料应从来源及工艺等生产环节进行严格控制,并应符合注射用的质量标准。
注射剂所用溶剂必须安全无害,并不得影响疗效和质量。
水溶性溶剂最常用的为注射用水,也可用0.9%氯化钠溶液或其他适宜的水溶液。
溶解注射用冻干制剂的无菌溶剂,通常也称注射用稀释剂,主要为灭菌注射用水、氯化钠注射液,应符合本版药典(二部)的规定。
其它药品标准卫生部颁药品标准(生化药品第一册)(38种)肝素钙注射液拼音名:Gansugai Zhusheye英文名:INJECTIO HEPARINI CALCII书页号:S1-15 标准编号:WS1-C3-0011-89 本品为肝素钙的灭菌水溶液。
其效价应为标示量的86.0~116.0%。
【性状】本品为无色或淡黄色的澄明液体。
【鉴别】照肝素钙项下的鉴别法试验,显相同的结果。
【检查】pH值应为5.0~8.0(中国药典1985年版二部附录33页)。
热原照肝素钙项下的方法检查,应符合规定。
其他应符合注射剂项下有关的各项规定(中国药典1985年版二部附录4 页)。
【效价测定】照肝素生物检定法测定(中国药典1985年版二部附录58页)。
【作用与用途】同肝素钙。
【用法与用量】皮下注射一次5,000~10,000单位一日一次。
【注意】用药期间应定时测定凝血时间。
有出血倾向及凝血机制障碍者禁用。
孕妇及产后妇女慎用。
【规格】(1) 1ml:5,000单位(2) 1ml:10,000单位(3) 2ml:10,000单位【贮藏】避光,在阴凉处保存。
有效期三年。
L-盐酸精氨酸拼音名:Yansuan Jing※ansuan英文名:L-ARGININI MONOHYDROCHLORIDUM书页号:S1-31 标准编号:WS1-C3-0022-89[C6H14N4O2²HCl=210.66] 本品为2-氨基-5-胍基戊酸盐酸盐。
按干燥品计算,含C6H14N4O2.HCl应为98.5~101.5%。
【性状】本品为白色结晶性粉末,水溶液显酸性反应。
本品在水中易溶,在乙醇中极微溶解。
比旋度取本品,精密称定,加盐酸溶液(9→100)制成每1ml中含30mg的溶液,依法测定(中国药典1985年版二部附录16页),按干燥品计算,比旋度应为+20.4°至+22.4°。
【鉴别】(1)取本品约2mg,加水2ml使溶解,加茚三酮约2mg,加热,溶液显蓝紫色。
附录生化药品文件编码(GHTU-UITID-GGBKT-POIU-WUUI-8968)生化药品附录第一章范围第一条本附录所指生化药品是指从动物的器官、组织、体液、分泌物中经前处理、提取、分离、纯化等制得的安全、有效、质量可控的药品。
主要包括:蛋白质、多肽、氨基酸及其衍生物、多糖、核苷酸及其衍生物、脂、酶及辅酶等(不包括生物制品附录所列产品)。
第二条本附录适用于原材料的前处理、提取、分离、纯化等原料(原液)及其制剂的制备和质量控制的全过程。
原材料的采集过程应符合国家相关规定,药品生产企业应监督控制其来源及质量。
第三条来源于人的组织、尿液的产品按照本附录执行。
第二章原则第四条应建立完善的质量管理体系,依据质量风险管理的原则,结合品种特点,明确从原材料采集至成品放行各阶段的质量管理责任,确保产品的安全有效、质量可控。
第五条生化药品具有以下特殊性,应对原材料的来源及质量、生产过程、中间产品的检验进行特殊控制:(一)生化药品的生产涉及器官、组织、体液、分泌物的提取、分离和纯化等过程,原材料本身具有不均一性。
(二)生化药品的质量控制通常采用生物分析技术,比理化测定具有更大的可变性。
(三)生产过程中的原材料和中间产品是污染微生物生长的良好培养基,原材料中的病原微生物对产品质量和生产环境存在较大风险。
第三章人员第六条从事生化药品生产、质量保证、质量控制、采购及其他相关人员(包括清洁、维修人员)均应根据其生产的产品和所从事的生产操作定期进行相关法律法规、专业知识、卫生和微生物学基础知识及安全防护要求等方面的培训及考核,并纳入个人培训档案。
第七条生产管理负责人、质量管理负责人和质量受权人应具有相应的专业知识(如微生物学、生物学、免疫学、药学、生物制药、生物化学等),并能够在生产、质量管理中切实履行职责。
从事供应商审计的人员,应了解动物种属、饲养、屠宰、检疫、采集及其原材料贮存运输等方面的相关知识,并能够在供应商管理和审核过程中有效履行其职责。
第八条应对所生产品种的生物安全进行评估,根据评估结果,生产、维修、检验的操作人员、管理人员应采取必要的生物安全防护措施。
第九条一般情况下,人员不应从原材料的前处理区域穿越到已经灭活产品、其他产品的处理区域。
如果不能避免这种穿越,必须基于质量风险管理原则采取防污染控制措施。
第四章厂房与设备第十条生化药品生产环境及厂房设施与设备不应对原材料、中间产品和成品造成污染;空气洁净度级别应与产品预定用途和生产操作相适应。
第十一条厂房应设有防止昆虫和其他动物等进入的设施。
特别是用于加工处理动物脏器、组织、体液或分泌物的生产操作区应配备有效的防虫防鼠措施,并评估其有效性。
第十二条原材料采集的厂房设施与设备应符合产品相应特性、卫生管理要求和国家相关规定,并与药品生产区域分开。
第十三条应结合产品潜在风险、不同生产阶段的工艺要求与特点,设置相应生产操作区域的环境控制要求,应尽可能降低产品(或原料)被微生物污染的风险。
第十四条在生产过程中应根据产品特性、工艺、预定用途和设备等因素,使用风险评估的手段,采取相应的预防差错、交叉污染、安全防护措施,如使用专用厂房和设备、阶段性生产方式、使用密闭系统等。
若使用敞口容器或设备操作时,应有避免污染的措施。
难以清洁的设备或部件应专用。
生化药品的去除/灭活病毒前的工艺步骤,不宜与其他动物源的药品共用设备和设施,不可避免时,应有适当的措施防止交叉污染。
第十五条原材料前处理应有专用区域,原料(原液)制备与制剂生产区域应严格分开。
原料(原液)制备和制剂生产的空调净化系统应分别独立设置。
第十六条原材料前处理和提取、纯化使用的设备、工器具、管道、阀门和容器具,包括取样器具应光洁、耐腐蚀、易清洗或消毒,并根据产品特性和过程控制要求进行有效的清洁或消毒处理。
第十七条洁净区内的冷冻或冷藏设施的使用、清洁、维护不应对洁净区环境造成污染。
第十八条用于物料及产品贮存的冷冻或冷藏设施应有预防突发事件发生的措施,避免物料及产品质量受到影响。
第五章病毒去除/灭活及验证第十九条应基于风险控制原则,结合品种特性和原材料来源,采用有效去除/灭活病毒工艺步骤和方法。
病毒去除/灭活的同时,不应对产品质量有不良影响。
第二十条去除/灭活病毒的生产工艺应有效并经验证,当生产工艺发生变更时,应重新评估验证的适用性,必要时应重新进行验证。
第二十一条病毒去除/灭活工艺的有效性验证可参照有关病毒去除/灭活技术方法和指导原则等相关规定。
第二十二条病毒去除/灭活方法的验证中的病毒挑战试验不得使用生产厂房设施和设备。
第二十三条病毒去除/灭活验证不能代替原材料及生产过程的质量管理要求。
第六章供应链管理第二十四条企业应针对生化药品供应链的广泛性和复杂性,基于质量风险管理原则建立有效的追溯系统和控制措施,应有文件明确供应链各环节的要求。
企业和供应商必须签订质量保证协议,明确相应的质量责任,要求供应商参照本附录管理。
第二十五条应对生化药品原材料的来源进行严格控制,原材料应来源于非疫区,并考虑来源地的流行病发生状况。
(一)原材料应来源于健康动物,纳入检验检疫管理的动物原材料均应来自经检疫合格的健康动物。
(二)来源于人的组织或尿液的,其采集应明确收集方法和要求。
(三)应定期收集动物来源区域疫情信息,评估质量风险。
当发现疫情风险时,应采取相应的质量控制措施。
(四)应根据原材料特性以及风险控制原则建立相应的追溯系统并记录。
第二十六条器官、组织、体液、分泌物等原材料采集单位应依法取得国家相关资质。
第二十七条质量管理部门应根据品种特点建立供应商质量管理档案,内容至少应包括:供应商的资质、规模、质量协议,原材料的动物来源、种属、年龄、采集部位及方法、采集后的保存方法与有效期等。
第二十八条应定期对原材料供应商进行现场审计。
重点考察供应链的各环节质量控制情况,确保供应商提供产品质量可控、稳定,并有质量审计报告。
第二十九条应对每批接收的原材料进行检查,并有相应记录:(一)供应商与质量管理部门批准的一致;(二)原材料附带检疫合格证明与货物一致;(三)包装标识与实物相符,标识内容应符合供应商档案中的相关内容;(四)外包装应完整无破损;(五)对贮存温度有特殊要求的,接收时应进行温度确认;运输全程的温度监控记录应完整可追溯,温度始终符合质量控制的要求。
第三十条原材料、中间品贮存和运输期间的包装材料或容器不应对产品质量产生影响,与其直接接触的包装材料应至少符合食品包装材料要求,供应商和材质应相对固定。
第三十一条冷库及冷链运输设备应经过确认。
原材料储存条件、储存期限、运输条件等经过确认,以保证产品质量。
第七章生产管理第三十二条应采取有效措施避免不同种属或同一种属的不同器官、组织、体液、分泌物在采集、转运及存放过程中的混淆、差错、污染、交叉污染。
第三十三条应对生化药品的原材料、中间品、原料(原液)进行批号或编号管理,以确保生产过程的可追溯性。
第三十四条应结合产品特性,尽量降低生产过程中微生物及其相关代谢物等的污染、交叉污染。
(一)应根据产品特性及贮存条件规定不同生产阶段的生产间隔时间,尽可能缩短不同生产阶段的时间间隔。
(二)中间产品、原料(原液)的储存时间应有明确规定并经验证。
(三)原材料、中间产品、原料(原液)不应反复冻融,必要时应经验证,以确保产品质量不受影响。
(四)生产结束后应在规定时限内对设备和容器进行清洁、消毒或灭菌。
(五)直接接触中间产品、原料(原液)的包装容器、设备清洁、消毒或灭菌后应避免再次污染。
提取、分离和纯化原料的各种设备、容器,不同产品和不同批次之间应进行清洁或消毒。
(六)同一设备通常不得用于不同产品或同一产品不同阶段的病毒去除/灭活操作。
如果使用同一设备,应采取适当的清洁或消毒措施,防止病毒通过设备或环境由前次操作带入后续纯化操作。
第三十五条生产中产生的副产品或废料应及时退出生产区域,防止污染生产环境及设备。
第三十六条应采取必要的措施,防止病毒去除或灭活后产品被污染;已经过病毒去除/灭活处理的产品与尚未处理的产品应有明显区分和标识,并应采用适当的方法防止混淆、差错。
第三十七条如有层析及超滤步骤,用于分离纯化的层析分离柱及超滤装置应专用。
同一层析分离柱及超滤装置不得应用于生产的不同阶段。
应有文件规定层析柱及超滤装置的可接受标准、操作条件、再生处理方法、使用寿命、清洗或消毒程序、过程监测参数等。
第三十八条生产所用的溶剂等需回收使用的,应制定回收操作规程及与其用途相适应的质量标准。
回收后的溶剂再使用不得对产品质量和安全性产生不利影响。
第八章质量管理第三十九条应按照《中华人民共和国药典》和国家食品药品监督管理部门批准的质量标准对生化药品原材料、辅料、中间品、原料(原液)及成品进行检验。
无法定标准的,企业应依据品种质量风险建立适宜的内控质量标准,必要时应考虑增加新鲜度、微生物限度、细菌内毒素或热原、异常毒性、降压物质、外源因子等检查项目。
第四十条生化药品的原材料来源应相对稳定,应明确动物的种属及器官组织。
必要时对原材料的动物种属进行鉴别(如PCR法等),取样应具有代表性。
第四十一条生化药品原材料的放行应包括完整的原材料追溯记录,纳入检验检疫管理的动物原材料均应来自经检疫合格的健康动物。
第四十二条原材料、辅料、中间品、原料(原液)的检验应在适当的生产阶段完成,当检验周期较长时,可先进行后续工艺生产,待检验合格后方可放行成品。
第四十三条必要时,中间品、原料(原液)应留样,以满足检测或中间控制确认的需要,留样数量应充足,并在适宜条件下贮存,便于质量追溯。
第九章术语第四十四条下列术语含义是:(一)原材料:用于药品生产的动物器官、组织、体液、分泌物以及人的组织、尿液等的起始物料,不包括辅料。
(二)原材料采集:从经过供应商审计合格的单位,收集动物的器官、组织、体液、分泌物及人组织、尿液的过程。
(三)前处理:对原材料进行的非药用部分的冻融、切割、拣选、洗涤以及后续进行的混合、离心、冻融等处理程序。
(四)中间品:原材料经前处理得到的产品,便于进行质量控制。
(五)原料(原液):中间品经提取、纯化、病毒去除/灭活,各项指标符合质量标准规定的产品。
(六)病毒去除/灭活:将病毒从产品中去除或灭活以保证安全的工艺过程。
(七)供应链:从原材料采集开始到制成中间产品的过程。