DOS图详解说明
- 格式:docx
- 大小:15.26 KB
- 文档页数:2

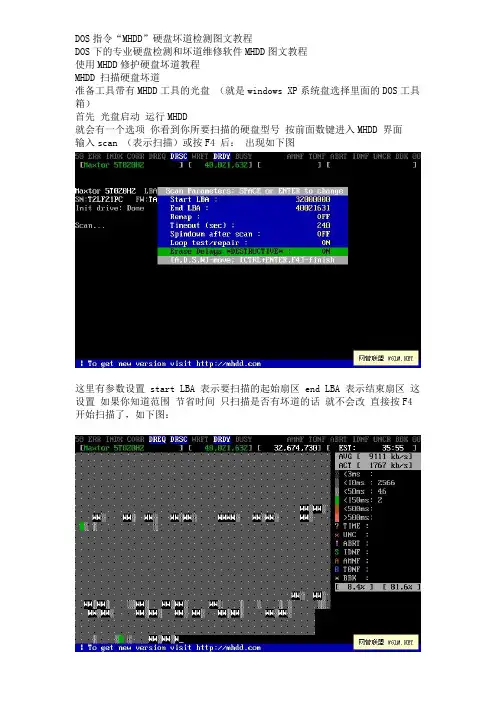
DOS指令“MHDD”硬盘坏道检测图文教程DOS下的专业硬盘检测和坏道维修软件MHDD图文教程使用MHDD修护硬盘坏道教程MHDD 扫描硬盘坏道准备工具带有MHDD工具的光盘(就是windows XP系统盘选择里面的DOS工具箱)首先光盘启动运行MHDD就会有一个选项你看到你所要扫描的硬盘型号按前面数键进入MHDD 界面输入scan (表示扫描)或按F4 后:出现如下图这里有参数设置 start LBA 表示要扫描的起始扇区 end LBA 表示结束扇区这设置如果你知道范围节省时间只扫描是否有坏道的话就不会改直接按F4 开始扫描了,如下图:下面说修复:当你扫描完后有坏道 MHDD扫描时候右下角会有数字那第一个就是扇区的起始最后一个就是坏道扇区结束修复和扫描一样按F4 进入参数设置界面但是要开启修复功能:如下图start LBA 填入你硬盘坏道的起始扇区 end LBA 填入你硬盘坏道结束扇区Remap 选为 ON 表示开启修复功能Timeout 为时间一盘是默认LOOP/TEST REPAIR 设置为ON 表示开启这里是反复擦写可以修复玩固环道ERASE 为OFF 表示关闭设置完成,按F4 开始修理了。
到这里扫描和修复就完了。
另外2种修复的方法,本人已经尝试过,非常管用!1.先按SHIFT+F3扫描硬盘连接并选择,按F4键,先用一般模式扫一遍,再用高级模式扫一变,具体方法是选择LBA模式,remap项OFF,Loop the test/repair 项OFF,其余项ON,选择完毕CTRL+ENTER执行扫描,扫描完毕,执行上面操作,选CHS模式,Loop the test/repair项OFF,Erase WAITs项OFF,其余项选ON,选择完毕,执行扫描,OK!!!大家切记不要直接用高级模式进行扫描,有些朋友为了急于求成,就直接用高级模式对硬盘进行操作,这种做法是错误的,如果直接用高级模式扫的话,一次好不了的话,那么MHDD对其就无可奈何了,要么认不到硬盘,要么坏道无法修复)修复成功率(1类坏道百分之九十以上,2类坏道百分之七十,3类坏道百分之十)2.下面这个方法,没耐心的朋友就不必看了。

DOS 下硬盘分区图详解硬盘分区并不复杂,只要你按照下面的图示步步为营,很快就能学会的!硬盘分区并不复杂,只要你按照下面的图示步步为营,很快就能学会的! 首先启动你的计算机,在BIOS 设置中,拨动启动顺序改为由光盘启动,然后把光盘放入光驱中接着就会出现三个选项:光盘放入光驱中接着就会出现三个选项:1。
START COMPUTER WITH CDROM SURPORT (不加载光驱);(不加载光驱); 2。
START COMPUTER WITHOUT CDROM SURPORT (加载光驱);(加载光驱); 3。
VIEW HELP FILES (显示帮助文件)(显示帮助文件)然后选2再回车出现A:\>A:\>敲入命令敲入命令fdisk 就回看见下图就回看见下图画面大意是说磁盘容量已经超过了512M 512M,为了充分发挥磁盘的性能,建议选用,为了充分发挥磁盘的性能,建议选用FAT32文件系统,输入“Y”键后按回车键。
文件系统,输入“Y”键后按回车键。
现在已经进入了Fdisk 的主画面,里面的选项虽然不多,但选项下面还有选项,操作时注意别搞混了。
操作时注意别搞混了。
图中选项解释:图中选项解释:1、创建DOS 分区或逻辑驱动器分区或逻辑驱动器2、设置活动分区、设置活动分区3、删除分区或逻辑驱动器、删除分区或逻辑驱动器4、显示分区信息、显示分区信息接下来将讲解如何创建新分区,接下来将讲解如何创建新分区,选择“1”后按回车键,画面显示如下:选择“1”后按回车键,画面显示如下:图中释义:图中释义: 1、创建主分区、创建主分区 2、创建扩展分区、创建扩展分区 3、创建逻辑分区、创建逻辑分区一般说来,硬盘分区遵循着“主分区→扩展分区→逻辑分区”的次序原则,硬盘分区遵循着“主分区→扩展分区→逻辑分区”的次序原则,而删而删除分区则与之相反。
除分区则与之相反。
一个硬盘可以划分多个主分区,一个硬盘可以划分多个主分区,一个硬盘可以划分多个主分区,但没必要划分那么多,但没必要划分那么多,但没必要划分那么多,一个一个足矣。

DOS 概述及入门——DOS 的特点在DOS 环境下,开机后,我们面对的不是桌面和图标,而是这样的电脑屏幕:这个C:\> 叫做提示符,这个闪动的横线叫做光标。
这样就表示电脑已经准备好,在等待我们给它下命令了。
我们现在所需要做的,就是对电脑发出命令,给电脑什么命令,电脑就会做什么。
那我们又怎么向电脑发布命令呢很简单。
比如,我们想看看现在几点钟了,就可以输入时间命令,敲入time 四个字母,它在英文中是“时间”的意思,再敲回车键加以确认,这时屏幕上就显示出当前时间。
再敲回车键,瞧!又出现提示符了,就可以输入下一条命令了。
如果想看日期,输入date,然后敲回车键确认。
这时屏幕上显示出当前日期。
再敲回车键就回到提示符下。
这就是DOS的命令输入方法,和Windows 95中用鼠标寻找图标来运行程序不同,DOS中是通过输入英文命令加回车键这种方式来执行程序的。
怎么样,和Windows 95 完全不一样吧在字符界面下,我们只能通过键盘输入字符来指挥电脑工作,电脑完成一个命令后,出现下一个提示符,我们就又可以给电脑下命令了。
注意,在DOS 下电脑一次只能做一件事,做完以后才能开始下一件事;而在95 下,我们可能让电脑同时干几件事,比如,我们可以一边拷贝文件,一边写文章,一边浏览In ternet。
所以人们把DOS称为单任务的操作系统,而把Windows 95 称为多任务的操作系统。
在DOS中,文件与目录是最重要的概念,这和95 一样,不同的是,目录”在95中叫做文件夹”。
如果我们想查看电脑中的文件,可以输入dir 命令,然后回车。
下面是我电脑屏幕上显示的结果。
如图所示,后面带有v dir>的是目录,没有的则是文件,这些目录里都分门别类地存放着许多不同用途的文件。
你看,第一排是DOS目录,它里面有许多DOS命令文件和一些辅助信息文件。
第二排是WINDOWS 目录,它里面包含着许多有关WINDOWS 程序的文件。


DOS处理:在dos计算文件夹中直接使用命令split_dos 处理得到DOS0 DOS1 DOS2 DOS3……分别对应总态密度,各个原子的态密度(一一对应POSCAR原子顺序)。
处理后的DOS0,DOS1….等文件的能量值是以费米能级作为能量参考点,DOS0的第一列数据是能量值,单位为eV,第二列数据是总态密度的值,单位为electrons. DOS1是第一个原子的分波态密度值,其中第一列数据是能量值,单位为eV。
第二三四列分别对应s, p, d, 态的分波态密度值,单位为State/eV.atom,其他文件类似DOS1.Sum_dos(sum_dos_np)使用说明:Sum_dos使用于自旋计算,sum_dos_np对应非自旋计算。
他们是把同一类原子的dos对应态密度值加起来!以GaN计算为例,若计算结果里有4个N 4个Ga(对应POSCAR里原子顺序为4个N在前,4个Ga在后),那么在dos计算出来split_dos处理后会出现DOS0 DOS1——DOS8,其中DOS1——DOS4对应的是4个N原子态密度,DOS5——DOS8对应是4个Ga态密度。
若为自旋计算,利用命令sum_dos 0 1 4处理得到DOS.SUM.1.to.4; DOS.SUM.5.to.8两个文件,分别为4个N(Ga)的s p d 态各自相加后的总值(里面每一列对应4个s态的和,4个p态的和,4个d态的和及s+p+d总和)。
非自旋计算利用sum_dos_np同样方式可得。
BAND画图处理:(以GaN为例)布里渊区路径选择为G-K-M-G如图所示:计算过程中两高对称点间插入30个点,首先测出G-K-M-G各段距离:在MS中选定一个线段左侧就能给出此线段距离如图所示:选择G-K段后此线段变为黄色,左侧显示出长度0.206490.同理可得到K-M M-G各段长度分为0.103245,0.179004。
然后新建EXCLE文件(此过程相当于把三段长度分别扩大30倍):如图所示:,然后左键点击0不放松下拉到0.20649 选定1,2行,变为,然后鼠标放到此黑框右下角位置(鼠标变为细黑体十字)点击下拉30行如图:第31行与30行同一个数如图:。

用第一原理计算软件开展的工作,分析结果主要是从以下三个方面进行定性/定量的讨论:1、电荷密度图(charge density);2、能带结构(Energy Band Structure);3、态密度(Density of States,简称DOS)。
电荷密度图是以图的形式出现在文章中,非常直观,因此对于一般的入门级研究人员来讲不会有任何的疑问。
唯一需要注意的就是这种分析的种种衍生形式,比如差分电荷密图(def-ormation charge density)和二次差分图difference charge density)等等,加自旋极化的工作还可能有自旋极化电荷密度图(spin-polarized charge density)。
所谓"差分"是指原子组成体系(团簇)之后电荷的重新分布,"二次"是指同一个体系化学成分或者几何构型改变之后电荷的重新分布,因此通过这种差分图可以很直观地看出体系中个原子的成键情况。
通过电荷聚集(accumulation)/损失(depletion)的具体空间分布,看成键的极性强弱;通过某格点附近的电荷分布形状判断成键的轨道(这个主要是对d轨道的分析,对于s或者p轨道的形状分析我还没有见过)。
分析总电荷密度图的方法类似,不过相对而言,这种图所携带的信息量较小。
能带结构分析现在在各个领域的第一原理计算工作中用得非常普遍了。
但是因为能带这个概念本身的抽象性,对于能带的分析是让初学者最感头痛的地方。
关于能带理论本身,我在这篇文章中不想涉及,这里只考虑已得到的能带,如何能从里面看出有用的信息。
首先当然可以看出这个体系是金属、半导体还是绝缘体。
判断的标准是看费米能级和导带(也即在高对称点附近近似成开口向上的抛物线形状的能带)是否相交,若相交,则为金属,否则为半导体或者绝缘体。
对于本征半导体,还可以看出是直接能隙还是间接能隙:如果导带的最低点和价带的最高点在同一个k点处,则为直接能隙,否则为间接能隙。

D O S图详解说明-CAL-FENGHAI-(2020YEAR-YICAI)_JINGBIAN用第一原理计算软件开展的工作,分析结果主要是从以下三个方面进行定性/定量的讨论:1、电荷密度图(charge density);2、能带结构(Energy Band Structure);3、态密度(Density of States,简称DOS)。
电荷密度图是以图的形式出现在文章中,非常直观,因此对于一般的入门级研究人员来讲不会有任何的疑问。
唯一需要注意的就是这种分析的种种衍生形式,比如差分电荷密图(def-ormation charge density)和二次差分图(difference charge density)等等,加自旋极化的工作还可能有自旋极化电荷密度图(spin-polarized charge density)。
所谓“差分”是指原子组成体系(团簇)之后电荷的重新分布,“二次”是指同一个体系化学成分或者几何构型改变之后电荷的重新分布,因此通过这种差分图可以很直观地看出体系中个原子的成键情况。
通过电荷聚集(accumulation)/损失(depletion)的具体空间分布,看成键的极性强弱;通过某格点附近的电荷分布形状判断成键的轨道(这个主要是对d轨道的分析,对于s或者p轨道的形状分析我还没有见过)。
分析总电荷密度图的方法类似,不过相对而言,这种图所携带的信息量较小。
能带结构分析现在在各个领域的第一原理计算工作中用得非常普遍了。
但是因为能带这个概念本身的抽象性,对于能带的分析是让初学者最感头痛的地方。
关于能带理论本身,我在这篇文章中不想涉及,这里只考虑已得到的能带,如何能从里面看出有用的信息。
首先当然可以看出这个体系是金属、半导体还是绝缘体。
判断的标准是看费米能级和导带(也即在高对称点附近近似成开口向上的抛物线形状的能带)是否相交,若相交,则为金属,否则为半导体或者绝缘体。
对于本征半导体,还可以看出是直接能隙还是间接能隙:如果导带的最低点和价带的最高点在同一个k点处,则为直接能隙,否则为间接能隙。

DOS与PDOS的介绍以及获取方法,注意事项dos与pdos要得到总的态密度dos和pdos按照侯老师的方法去搞,先优化、静态,再算出来dos,获得doscar.接着就要用小程序split_dos对doscar进行分割。
可以得到dos0、dos1、dos2............其中dos0是总的态密度,dos1、dos2............分别是第一、第二............个原子的分波态密度。
dos1就是第一个原子的分波态密度值,其中第一列就是能量,第二、三、四列数据分别对应于s、p、d态的分波态密度值。
split_dos做出来的tdos,费米能级就在0ev处,意思就是说将费米能3.1653归零处理,其他的能量其实都是相对于3.1653ev的相对值推论溶解稳定性先看结合能,然后再分析电荷转移情况,再分析pdos以判断来源。
稳定与否应该可以通过对应吸附原子的pdos往低能移动来解释楼主可以结合态密度、分波态密度和电荷密度以及溶解能够四项去综合实地考察费米能级附近态密度越阔,表明成键越平衡不确认,等待核实总的态密度为什么和分波态密度不相等?这与排序态密度的时候所对原子空间的分割方法有关~只要横坐标的能量对应上了就好了。
纵坐标累加不等tdos是完全在正常的。
总态密度的单位是电子/单胞体积分波态密度的单位是电子/原子数分态dos强度和总dos强度不成正比就是很正常的,关键看看峰值的能量范围若想和总的dos对应出来,且分态的dos也就是只图画单原子的即可,没有必要加和。
dos图中怎么判断非金属性,半金属性,金属性只看看整体,如果单个元素的分波态密度越过费米能级,那整体必须就是金属性或者有可能就是半金属性的dos与bandstructure都能看出导体、半导体、半金属等,在费米面处存在能隙的,则为非金属或半导体。
能带结构的分析:推论的标准就是看看费米能级和导带(也即为在低对称点附近对数成开口向上的抛物线形状的能带)与否平行,若平行,则为金属,否则为半导体或者绝缘体。

一. 命令行简介命令行就是在Windows操作系统中打开DOS窗口,以字符串的形式执行Windows管理程序。
在这里,先解释什么是DOS?DOS——Disk Operation System 磁盘操作系统目前我们常用的操作系统有windows 9x/Me,NT,2000等,都是可视化的界面。
在这些系统之前的人们使用的操作系统是DOS系统。
DOS系统目前已经没有什么人使用了,但是dos命令却依然存在于我们使用的windows系统之中。
大部分的DOS命令都已经在Windows里变成了可视化的界面,但是有一些高级的DOS命令还是要在DOS环境下来执行。
所以学习命令行对于我们熟练操作Windows系统是很有必要的。
不同的操作系统要用不同的命令进入命令行界面。
在Win9x/Me的开始菜单中的运行程序中键入"command"命令,可进入命令行界面。
在Win2000/NT的开始菜单中的运行程序中键入"cmd"命令,可进入命令行界面。
下面我用讲到的DOS命令都可以在Windows Me操作系统中执行。
那么,我们如何进入命令行窗口?开始——〉运行——〉键入command命令——〉回车进入了命令行操作界面(DOS窗口),在DOS窗口中只能用键盘来操作。
如下所示:二.符号约定为了便于说明格式,这里我们使用了一些符号约定,它们是通用的:C: 盘符Path 路径Filename 文件名.ext 扩展名Filespec 文件标识符[ ] 方括号中的项目是可选项,用户可以根根据需要不输入这些内容{ } 大括号表示其中的项目必选一项| 竖线表示两侧的内容可取其一…表示可重复项二.三.命令行的输入方法在DOS窗口中通过输入英文命令加回车键这种方式来执行程序。
四.内部命令和外部命令命令行程序分为内部命令和外部命令,内部命令是随装入内存的,而外部命令是一条一条单独的可执行文件。
内部命令都集中在根目录下的文件里,电脑每次启动时都会将这个文件读入内存,也就是说在电脑运行时,这些内部命令都驻留在内存中,用dir命令是看不到这些内部命令的。

MHDD 图解教程1、MHDD是俄罗斯Maysoft公司出品的专业硬盘工具软件,具有很多其他硬盘工具软件所无法比拟的强大功能,它分为免费版和收费的完整版,本文介绍的是免费版的详细用法。
这是一个G表级的软件,他将扫描到的坏道屏蔽到磁盘的G表中。
(小知识:每一个刚出厂的新硬盘都或多或少的存在坏道,只不过他们被厂家隐藏在P表和G表中,我们用一般的软件访问不到他。
G表,又称用户级列表,大约能存放几百个到一千左右的坏道;P表,又称工厂级列表,能存放4000左右的坏道或更多。
)由于它扫描硬盘的速度非常快,已成为许多人检测硬盘的首选软件。
2、此软件的特点:不依赖主板BIOS,支持热插拔。
MHDD可以不依赖于主板BIOS直接访问IDE口,可以访问128G的超大容量硬盘(可访问的扇区范围从512到137438953472),即使你用的是286电脑,无需BIOS支持,也无需任何中断支持.热插拔的顺序要记清楚:插的时候,先插数据线,再插电源线。
拔的时候,先拔电源线,再拔数据线。
但我劝你不熟练最好不要热插拔,以免你不小心烧了硬盘赖我。
3、MHDD最好在纯DOS环境下运行;但要注意尽量不要使用原装Intel品牌主板;4、不要在要检测的硬盘中运行MHDD;5、MHDD在运行时需要记录数据,因此不能在被写保护了的存储设备中运行(比如写保护的软盘、光盘等)下面,我们在DOS下运行MHDD29:输入命令MHDD29,按回车,出现主界面:主界面列出了MHDD的所有命令,下面我们主要讲解MHDD的几个常用命令:PORT;ID ;SCA N;HPA;RHPA;NHPA;PWD ;UNLOCK ;DISPWD ;ERASE ;AERASE ;STOP。
首先输入命令PORT(热键是:SHIFT+F3),按回车。
这个命令的意思是扫描IDE口上的所有硬盘。
好了,现在看到有两个硬盘,一个是西数40G,一个是迈拓2G。
(说明:1、2是接在IDE1口上的主从硬盘,3、4是接在IDE2口上的主从硬盘,5是接在PC3000卡上的。
DOS:是态密度,是某个能量处的态的数目。
这个数目包含了各个分子轨道的贡献(包括占据轨道和未占据轨道,占据轨道的贡献一般在能量较低的位置,未占据轨道的贡献一般在能量较高处)。
因为DOS对能量空间积分后的数值等于计算中用的基函数的数量。
而基函数的数量等于计算出来的本征波函数(或分子轨道,这样称呼有时候不合适,但是为了方便还是这样叫吧)。
PDOS:是把DOS投影到各个基函数上。
要想计算一个原子轨道的PDOS,只需要将构成这个原子轨道的各个基函数的PDOS求和。
要想求某个原子对PDOS 的贡献,只需要将该原子在计算的时候使用的原子轨道求和就可以了。
既然原子轨道是定域的,也就是说以原子核为中心的,所以对某一个原子求PDOS实际上就算出LDOS了,LDOS是表现在某个能量范围内,态密度在空间的分布。
是表现在某一个能量范围内,原子上某个原子轨道上DOS的分布。
侧重点不同。
画PDOS实际上就是指明了空间位置了相当于研究的是LDOS的某一个空间点上的情况。
当然PDOS可以分析某一元素对DOS的贡献,这个就不是局域的了。
能带:金属、半导体和绝缘体的区别。
能带可分为价带、禁带和导带三部分,倒带和价带之间的空隙称为能隙,基本概念如图所示:如何能隙很小或为0 ,则固体为金属材料,在室温下电子很容易获得能量而跳跃至导带而导电;而绝缘材料则因为能隙很大(通常大于9电子伏特),电子很难跳跃至传导带,所以无法导电。
一般半导体材料的能隙约为1至3电子伏特,介于导体和绝缘体之间。
因此只要给予适当条件的能量激发,或是改变其能隙之间距,此材料就能导电。
能带用来定性地阐明了晶体中电子运动的普遍特点。
价带(valence band),或称价电带,通常指绝对零度时,固体材料里电子的最高能量。
在导带(conduction band)中,电子的能量范围高于价带,而所有在传导带中的电子均可经由外在的电场加速而形成电流。
对与半导体以及绝缘体而言,价带的上方有一个能隙(band gap),能隙上方的能带则是传导带,电子进入传导带后才能在固体材料内自由移动,形成电流。
处理:DOS在计算文件夹中直接使用命令处理得到DOS0DOS1split_dosdos分别对应总态密度,各个原子的态密度(一一对DOS3……DOS2应原子顺序)。
处理后的,等文件的能量值DOS1….DOS0POSCAR 是以费米能级作为能量参考点,的第一列数据是能量值,单DOS0位为第二列数据是总态密度的值,单位为是第electrons.,DOS1eV 一个原子的分波态密度值,其中第一列数据是能量值,单位为。
eV 第二三四列分别对应态的分波态密度值,单位为d,s,p,其他文件类似DOS1.State/eV.atom,使用说明:Sum_dos(sum_dos_np)使用于自旋计算,对应非自旋计算。
他们是把Sum_dos sum_dos_np同一类原子的对应态密度值加起来!以计算为例,若计算GaN dos结果里有个个(对应里原子顺序为个在前,N N44Ga4 POSCAR,那么在计算出来处理后会出现个在后)split_dos Ga DOS04dos ——,其中——对应的是个原子态密4DOS1NDOS4DOS1DOS8度,——对应是个态密度。
若为自旋计算,利用4GaDOS5DOS8命令处理得到DOS.SUM.1.to.4;104DOS.SUM.5.to.8sum_dos态各自相加后的总值两个文件,分别为个()的sGa4N pd(里面每一列对应个态的和,个态的和,个态的和及p4d s44。
非自旋计算利用同样方式可得。
和)总s+p+d sum_dos_np (以为例)画图处理:BAND GaN布里渊区路径选择为如图所示:G-K-M-G计算过程中两高对称点间插入个点,首先测出各段距G-K-M-G30离:在中选定一个线段左侧就能给出此线段距离如图所示:MS选择段后此线段变为黄色,左侧显示出长度同理可得0.206490.G-K 到各段长度分为,。
0.1790040.103245M-G K-M然后新建文件(此过程相当于把三段长度分别扩大:倍)30EXCLE如图所示:,然后左键点击不放松下拉到0.206490选定,行,变为,然后鼠标放到此黑框右下角位置21(鼠标变为细黑体十字)点击下拉行如图:30第行与行同一个数如图:。
用第一原理计算软件开展的工作,分析结果主要是从以下三个方面进行定性/定量的讨论:
1、电荷密度图(charge density);
2、能带结构(Energy Band Structure);
3、态密度(Density of States,简称DOS)。
电荷密度图是以图的形式出现在文章中,非常直观,因此对于一般的入门级研究人员来讲不会有任何的疑问。
唯一需要注意的就是这种分析的种种衍生形式,比如差分电荷密图(def-ormation charge density)和二次差分图(difference charge density)等等,加自旋极化的工作还可能有自旋极化电荷密度图(spin-polarized charge density)。
所谓“差分”是指原子组成体系(团簇)之后电荷的重新分布,“二次”是指同一个体系化学成分或者几何构型改变之后电荷的重新分布,因此通过这种差分图可以很直观地看出体系中个原子的成键情况。
通过电荷聚集(accumulation)/损失(depletion)的具体空间分布,看成键的极性强弱;通过某格点附近的电荷分布形状判断成键的轨道(这个主要是对d轨道的分析,对于s或者p轨道的形状分析我还没有见过)。
分析总电荷密度图的方法类似,不过相对而言,这种图所携带的信息量较小。
能带结构分析现在在各个领域的第一原理计算工作中用得非常普遍了。
但是因为能带这个概念本身的抽象性,对于能带的分析是让初学者最感头痛的地方。
关于能带理论本身,我在这篇文章中不想涉及,这里只考虑已得到的能带,如何能从里面看出有用的信息。
首先当然可以看出这个体系是金属、半导体还是绝缘体。
判断的标准是看费米能级和导带(也即在高对称点附近近似成开口向上的抛物线形状的能带)是否相交,若相交,则为金属,否则为半导体或者绝缘体。
对于本征半导体,还可以看出是直接能隙还是间接能隙:如果导带的最低点和价带的最高点在同一个k点处,则为直接能隙,否则为间接能隙。
在具体工作中,情况要复杂得多,而且各种领域中感兴趣的方面彼此相差很大,分析不可能像上述分析一样直观和普适。
不过仍然可以总结出一些经验性的规律来。
主要有以下几点:1)因为目前的计算大多采用超单胞(supercell)的形式,在一个单胞里有几十个原子以及上百个电子,所以得到的能带图往往在远低于费米能级处非常平坦,也非常密集。
原则上讲,这个区域的能带并不具备多大的解说/阅读价值。
因此,不要被这种现象吓住,一般的工作中,我们主要关心的还是费米能级附近的能带形状。
2)能带的宽窄在能带的分析中占据很重要的位置。
能带越宽,也即在能带图中的起伏越大,说明处于这个带中的电子有效质量越小、非局域(non-local)的程度越大、组成这条能带的原子轨道扩展性越强。
如果形状近似于抛物线形状,一般而言会被冠以类sp带(sp-like band)之名。
反之,一条比较窄的能带表明对应于这条能带的本征态主要是由局域于某个格点的原子轨道组成,这条带上的电子局域性非常强,有效质量相对较大。
3)如果体系为掺杂的非本征半导体,注意与本征半导体的能带结构图进行对比,一般而言在能隙处会出现一条新的、比较窄的能带。
这就是通常所谓的杂质态(doping state),或者按照掺杂半导体的类型称为受主态或者施主态。
4)关于自旋极化的能带,一般是画出两幅图:majority spin和minority spin。
经典的说,分别代表自旋向上和自旋向下的轨道所组成的能带结构。
注意它们在费米能级处的差异。
如果费米能级与majority spin的能带图相交而处于minority spin的能隙中,则此体系具有明显的自旋极化现象,而该体系也可称之为半金属
(half metal)。
因为majority spin与费米能级相交的能带主要由杂质原子轨道组成,所以也可以此为出发点讨论杂质的磁性特征。
5)做界面问题时,衬底材料的能带图显得非常重要,各高对称点之间有可能出现不同的情况。
具体地说,在某两点之间,费米能级与能带相交;而在另外的k 的区间上,费米能级正好处在导带和价带之间。
这样,衬底材料就呈现出各项异性:对于前者,呈现金属性,而对于后者,呈现绝缘性。
因此,有的工作是通过某种材料的能带图而选择不同的面作为生长面。
具体的分析应该结合试验结果给出。
(如果我没记错的话,物理所薛其坤研究员曾经分析过$\beta$-Fe的(100)和(111)面对应的能带。
有兴趣的读者可进一步查阅资料。
)
原则上讲,态密度可以作为能带结构的一个可视化结果。
很多分析和能带的分析结果可以一一对应,很多术语也和能带分析相通。
但是因为它更直观,因此在结果讨论中用得比能带分析更广泛一些。
简要总结分析要点如下:
1)在整个能量区间之内分布较为平均、没有局域尖峰的DOS,对应的是类sp 带,表明电子的非局域化性质很强。
相反,对于一般的过渡金属而言,d轨道的DOS一般是一个很大的尖峰,说明d电子相对比较局域,相应的能带也比较窄。
2)从DOS图也可分析能隙特性:若费米能级处于DOS值为零的区间中,说明该体系是半导体或绝缘体;若有分波DOS跨过费米能级,则该体系是金属。
此外,可以画出分波(PDOS)和局域(LDOS)两种态密度,更加细致的研究在各点处的分波成键情况。
3)从DOS图中还可引入“赝能隙”(pseudogap)的概念。
也即在费米能级两侧分别有两个尖峰。
而两个尖峰之间的DOS并不为零。
赝能隙直接反映了该体系成键的共价性的强弱:越宽,说明共价性越强。
如果分析的是局域态密度(LDOS),那么赝能隙反映的则是相邻两个原子成键的强弱:赝能隙越宽,说明两个原子成键越强。
上述分析的理论基础可从紧束缚理论出发得到解释:实际上,可以认为赝能隙的宽度直接和Hamiltonian矩阵的非对角元相关,彼此间成单调递增的函数关系。
4)对于自旋极化的体系,与能带分析类似,也应该将majority spin和minority spin 分别画出,若费米能级与majority的DOS相交而处于minority的DOS的能隙之中,可以说明该体系的自旋极化。