常见载体引物与序列
- 格式:xlsx
- 大小:16.51 KB
- 文档页数:5
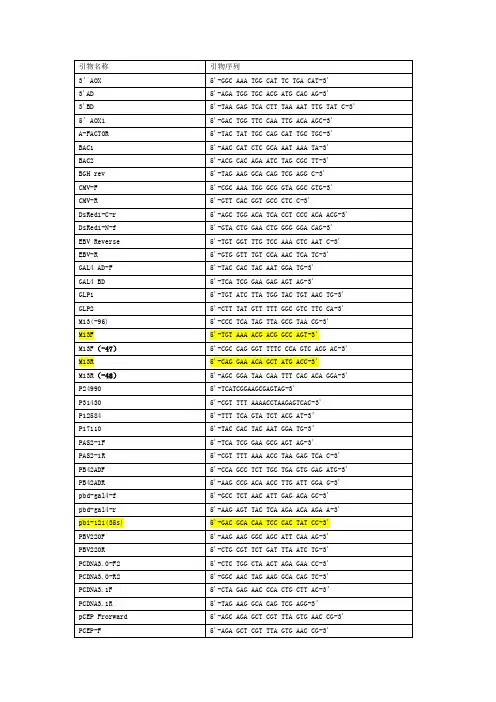

引物的应用常识引物的原理引物是短的寡核苷酸片段,充当DNA复制的起点。
因为几乎所有DNA聚合酶都不能从头合成,所以它们需要一个3’-羟基作为DNA合成的起始点。
这个3’-羟基由相配的引物提供。
在体内,由于DNA聚合酶的忠实性,不能从头合成DNA,因此只能由RNA聚合酶(称为引物酶)生成,采用RNA引物来延伸,在延伸过程中,RNA引物降解并由DNA取代。
在体外PCR反应中所用到的DNA引物,是根据不同的要求及模板序列设计,然后用化学法人工合成的,与模板形成双链后在DNA聚合酶的作用下就可以继续链的延伸;对于大多数PCR反应,决定整个反应成功与否的最重要因素是引物的序列和质量。
1. 不同实验要求的引物选择在开始设计引物之前,必须弄清以下几点:(1)明确PCR的目的(例如克隆、SNP检测、定量检测等)(2)确定样品材料(基因组DNA、RNA、微小RNA)(3)确定PCR的类型(普通的、定量PCR、RT-PCR、长片段PCR),在查找序列的时候还需要考虑可能存在的问题(如假基因等)2.引物设计的重要因素有一些不同的软件工具可用于引物设计和引物分析。
引物设计的软件如Oligo 6.22 ,Premier 5.0,Primer Express 3。
引物分析常用Primer 5,Oligo 6.22,Primer-Blast。
目前生工生物给客户提供的引物设计服务引物用的是在线软件Primer 3 plus,•引物长度和专一性常见的引物长度为18-30个碱基。
短的引物(≤15碱基)能非常高效地结合, 但是它们的专一性不够。
较长的引物能提高专一性,然而退火效率低,从而导致PCR产量低下。
同时应避免编码单一序列和重复序列的引物。
•平衡GC含量,避免GC-和AT-富集区域引物的GC含量应介于40%~60%之间。
应避免聚-(dC)-或聚(dG)-区域,因为它们会降低退火反应的专一性。
聚-(dA)-和聚(dT)-也应避免,因为这样会形成不稳定的引物-模板复合物,从而降低扩增效率。

真核细胞常见表达载体1. pCMVp-NEO-BAN载体特点: 该真核细胞表达载体分子量为6600碱基对,主要由CMVp启动子、兔β-球蛋白基因内含子、聚腺嘌呤、氨青霉素抗性基因和抗neo基因以及pBR322骨架构成,在大多数真核细胞内都能高水平稳定地表达外源目的基因。
更重要的是,由于该真核细胞表达载体中抗neo基因存在,转染细胞后,用G418筛选,可建立稳定的、高表达目的基因的细胞株。
插入外源基因的克隆位点包括Sal1、BamH1和EcoR1位点。
注意在此载体中有二个EcoR1位点存在。
2. pEGFP, 增强型绦色荧光蛋白表达载体(Enhanced Fluorecent Protein Vector特点: pEGFP表达载体中含有绿色荧光蛋白,在PCMV启动子驱动下,在真核细胞中高水平表达。
载体骨架中的SV40 origin使该载体在任何表达SV40 T 抗原的真核细胞内进行复制。
Neo抗性盒由SV40早期启动子、Tn5的neomycin/kanamycin抗性基因以及HSV-TK基因的聚腺嘌呤信号组成,能应用G418筛选稳定转染的真核细胞株。
此外,载体中的pUC origin 能保证该载体在大肠杆菌中的复制,而位于此表达盒上游的细菌启动子能驱动kanamycin抗性基因在大肠杆菌中的表达。
用途: 该表达载体EGFP上游有Nde1、Eco47111和Age1克隆位点,将外源基因扦入这些位点,将合成外源基因和EGFP的融合基因。
借此可确定外源基因在细胞内的表达和/或组织中的定位。
亦可用于检测克隆的启动子活性(取代CMV启动子,Acet1-Nhe1。
Excitation maximum = 488 nm; Emission maximum = 507图示为启动子分泌信号肽和多克隆位点区域:Ase1.pCMV…ccg cta gcg cta ccg gtc gcc acc atg- .EGFP…BamH1…SV40 poly A+Nhe1 Age13. pEGFT-Actin, 增强型绿色荧光蛋白/人肌动蛋白表达载体特点: pEGFP-Actin表达载体中含有绿色荧光蛋白和人胞浆β-肌动蛋白基因,在PCMV启动子驱动下,在真核细胞中高水平表达。

引物设计和载体构建知识点引物设计和载体构建是分子生物学中重要且基础的实验技术,它们在基因克隆、基因组编辑等方面起到了不可替代的作用。
本文将对引物设计和载体构建的相关知识点进行介绍。
一、引物设计引物是指在PCR等实验中用于扩增特定DNA片段的短寡核苷酸序列。
一个好的引物设计能够确保PCR扩增的特异性和高效性。
1. 引物长度引物的长度通常在18-30个碱基对之间,过短的引物可能导致扩增非特异性产物,而过长的引物则可能降低扩增效率。
2. 引物序列引物序列应该与目标DNA片段的序列互补,并且避免二聚体和头部稳定性等问题。
同时,还要注意避免引物内部和引物之间的序列相互互补。
3. 引物的Tm值引物的Tm值是指引物与目标DNA片段结合的解离温度。
引物的Tm值应该在50-65摄氏度之间,以确保引物的特异性和高效性。
4. 引物的GC含量引物的GC含量直接影响引物的稳定性和特异性。
过高或过低的GC含量可能导致非特异性扩增产物的生成。
通常情况下,GC含量在40-60%之间较为理想。
二、载体构建载体是指在基因工程中用于携带外源DNA片段并将其导入到宿主细胞中的分子。
载体构建是基因克隆和基因组编辑等实验的基础。
1. 选择合适的载体选择合适的载体是载体构建的第一步。
常用的载体包括质粒、病毒和噬菌体等。
根据实验需要选择合适的载体,例如质粒常用于DNA片段的克隆和表达,而病毒则常用于基因传递和基因治疗。
2. 载体的线性化和限制酶切线性化载体有助于DNA片段的插入和连接。
通过使用适当的限制酶对载体进行切割,生成具有完整黏性末端的线性载体。
3. DNA片段的连接将目标DNA片段与线性载体进行连接,一般采用DNA连接酶或者DNA ligation kit。
连接后的载体能够稳定地携带外源DNA片段。
4. 载体的转化和筛选将构建好的载体导入到宿主细胞中,通过培养基中的选择性抗生素或者其他筛选方法选择具有外源DNA片段的转化子。
总结:引物设计和载体构建是分子生物学实验中的重要环节,它们对于基因克隆、基因组编辑等研究具有重要意义。

常用pGEX载体图谱Rosetta系列的表达菌株可以提供T7 RNA聚合酶,它能表达PET系列载体上的外源基因。
pGEX系列载体上的外源基因不需要T7 RNA 聚合酶,普通的大肠杆菌经IPTG诱导即可表达Tac启动子是一组由Lac和trp启动子人工构建的杂合启动子,受Lac阻遏蛋白的负调节,它的启动能力比Lac和trp都强。
其中Tac 1是由Trp启动子的-35区加上一个合成的46 bp DNA片段(包括Pribnow 盒)和Lac操纵基因构成,Tac 12是由Trp的启动子-35区和Lac 启动子的-10区,加上Lac操纵子中的操纵基因部分和SD序列融合而成蛋白标签:A myc tag is a polypeptide proteintag derived from the c-myc gene product that can be added to a proteinusing recombinant DNA technology. It can be used for affinity chromatography, then used to separate recombinant, overexpressed protein from wild type protein expressed by the host organism. Itcan also be used in the isolation of protein complexes with multiple subunits.A myc tag can be used in many different assays that require recognition byan antibody. If there is no antibody against the studied protein, adding a myc-tag allows one to follow the protein with an antibody against the Myc epitope. Examples are cellular localization studies by immunofluorescence or detectionby Western blotting.The peptide sequence of the myc-tag is (in 1- and 3-letter codes, respectively):N-EQKLISEEDL-C,N-Glu-Gln-Lys-Leu-Ile-Ser-Glu-Glu-Asp-Leu -C, where N stands for Amino-terminus and C stands for Carboxy terminus. The tag is approximately 1202 Daltons in atomic mass and has 10 amino acids.It can be fused to the C-terminus andthe N-terminus of a protein. It is advisablenot to fuse the tag directly behind the signal peptide of a secretory protein, since it can interfere with translocation intothe secretory pathway.A monoclonal antibody against themyc epitope, named 9E10, is available from the non-commercial Developmental Studies Hybridoma BankpGEX4T1载体基本信息出品公司: GE别名: pGEX-4T-1, pGEX4T1, pGEX 4T 1质粒类型: 大肠杆菌蛋白表达载体表达水平: 高拷贝启动子: Tac克隆方法: 多克隆位点,限制性内切酶载体大小: 4969 bp5' 测序引物及序列: pGEX5': GGGCTGGCAAGCCACGTTTGGTG3' 测序引物及序列: pGEX3': CCGGGAGCTGCATGTGTCAGAGG载体标签: N-GST载体抗性: Ampicillin 氨苄备注: 复制子是pMB1产品目录号: 27-4580-01稳定性: 瞬时表达 Transient组成型: 诱导表达病毒/非病毒: 非病毒pGEX4T1载体质粒图谱和多克隆位点信息原核生物DNA复制起始点,是DNA链上独特的具有起始DNA复制功能的碱基序列。

高二生物选修三知识点pcr引物PCR引物是在聚合酶链式反应(PCR)中起着重要作用的短片段DNA序列。
它们的主要功能是在PCR反应中选择性地扩增目标DNA序列。
PCR引物的设计对于PCR反应的成功与否至关重要。
本文将介绍高二生物选修三中关于PCR引物的知识点。
首先,PCR引物的选择需要考虑以下几个要素:引物长度、碱基组成、配对温度和目标DNA序列的特点。
在选择引物长度时,一般建议引物长度在18-30个碱基对之间,过短的引物可能导致扩增产物过多,过长的引物则可能增加非特异性扩增的风险。
其次,DNA的碱基组成也是选择引物的重要因素之一。
引物的碱基组成应尽量避免多个相邻的鳕嘧啶或腺嘌呤碱基,因为它们容易形成二聚体结构,从而降低PCR反应的效率。
此外,引物的GC含量一般控制在40-60%之间,过高或过低的GC含量都可能导致PCR反应的失败。
再次,引物的配对温度是指引物与目标DNA序列结合的温度。
通常,引物的配对温度应该在5-10摄氏度低于目标DNA序列的Tm(熔解温度)。
Tm是指DNA双链解链的温度,可以通过计算DNA序列的碱基组成来估算。
最后,PCR引物的设计还需要考虑目标DNA序列的特点。
例如,如果目标DNA序列中含有重复序列或者结构变异区域,需要避免引物与这些区域的匹配,以免影响PCR反应的特异性。
除了以上几点,还有一些常见的PCR引物设计策略。
例如,引物的末端可以添加一段序列,以便引物与载体或标签结合。
此外,在多重引物PCR反应中,引物的选择需要确保每个引物对应的目标区域不重叠,以免干扰扩增产物的解析。
总结来说,PCR引物的设计对于PCR反应的成功至关重要。
选择合适的引物可以提高PCR的特异性和效率,从而确保准确可靠的扩增目标DNA序列。
高二生物选修三中关于PCR引物的知识点主要包括引物长度、碱基组成、配对温度和目标DNA序列的特点等。
通过合理设计PCR引物,可以在实验中取得准确的PCR扩增结果,并在分子生物学研究中发挥重要作用。
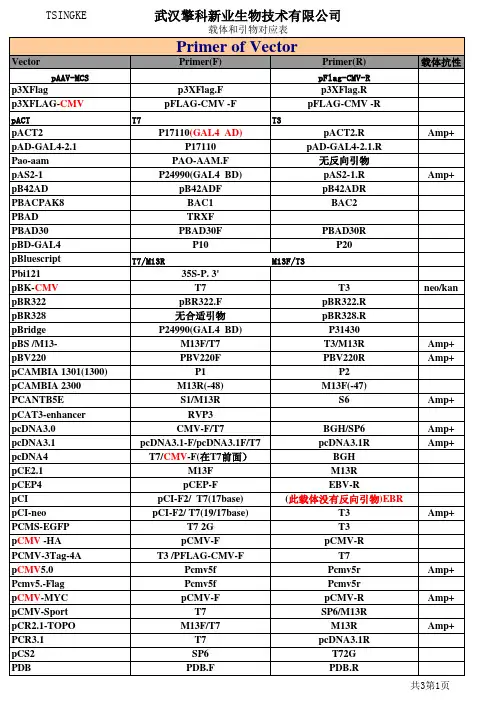

目的基因与载体连接的方法基因工程技术是通过将外源基因导入到宿主细胞中,以改变宿主细胞的遗传特征。
在基因工程中,目的基因常常需要连接到载体上才能被导入到宿主细胞中。
下面将介绍几种常见的目的基因与载体连接的方法。
1. 限制性内切酶切割连接法限制性内切酶是一类能够识别特定的DNA序列并切割DNA链的酶。
利用限制性内切酶的特异性切割性质,可以在目的基因和载体的DNA链上选择合适的切割位点。
切割后的DNA链上会留下一段不互补的单链,这个单链被称为粘性末端。
将目的基因和载体分别经过限制性内切酶切割后,可以利用这种粘性末端相互结合,形成互补碱基配对,然后使用DNA连接酶将其连接起来。
2. PCR扩增连接法PCR (聚合酶链反应) 是一种常用的基因扩增技术。
利用PCR技术,可以通过引物扩增得到目的基因和载体的DNA片段。
设计引物时,在其序列的3'端加入适当的限制性内切酶切割位点,然后通过PCR扩增获得含有限制性内切酶切割位点的DNA片段。
然后,将PCR产物和载体经过限制性内切酶切割后,利用DNA 连接酶将它们连接起来。
3. 转座子连接法转座子是一种能够在基因组内移动的DNA序列。
转座子连接法的原理是利用转座酶催化转座子在目的基因和载体之间的移动。
首先,将转座子序列插入到载体的DNA链上,然后通过反转录酶合成一个带有转座子的RNA分子。
此RNA 分子与目的基因连接后,通过转座酶的作用,将整个RNA-目的基因-转座子复合物插入到目的细胞的基因组中。
4. DNA连接酶片段连接法DNA连接酶片段连接法利用DNA连接酶催化DNA片段之间的连接。
该方法使用DNA连接酶作为催化剂将目的基因和载体的DNA片段连接起来。
此方法需要目的基因和载体之间有一段互补的序列,以便DNA连接酶能够催化它们连接起来。
5. Ligation-Independent Cloning (LIC)连接法LIC连接法是一种无连接酶的连接方法。
该方法不需要限制性内切酶切割,也无需互补的粘性末端。

pBI221-‐GFP编号 载体名称北京华越洋生物VECT0320 pBI221-‐GFPpBI221-‐GFP载体基本信息出品公司: -‐-‐载体名称: pBI221-‐GFP质粒类型: 植物双元表达载体高拷贝/低拷贝: -‐-‐启动子: -‐-‐克隆方法: 多克隆位点,限制性内切酶载体大小: -‐-‐5' 测序引物及序列: -‐-‐3' 测序引物及序列: -‐-‐载体标签: GFP载体抗性: -‐-‐筛选标记: -‐-‐备注: -‐-‐产品目录号: -‐-‐稳定性: -‐-‐组成型: -‐-‐病毒/非病毒: -‐-‐载体质粒图谱和多克隆位点信息其他植物载体质粒:pBI101 pDF15pBI121 pEarleyGate 100 pBI221 pEarleyGate 101 pBI221-‐GFP pEarleyGate 102 pBin19 pEarleyGate 103pBINPLUS pEarleyGate 104 pCambia0105.1R pEarleyGate 201 pCambia0305.1 pEarleyGate 202 pCambia0305.2 pEarleyGate 203 pCambia0380 pEarleyGate 204 pCambia0390 pEarleyGate 205 pCambia1105.1 pEarleyGate 301 pCambia1105.1R pEarleyGate 302 pCambia1200 pEarleyGate 303 pCambia1201 pEarleyGate 304 pCambia1281Z pFGC5941 pCambia1291Z pGA643 pCambia1300 pGreen pCambia1300GFP pGreen 0029 pCambia1301 pGreen0029 pCambia1302 pGreen029 pCambia1303 pGreenII pCambia1304 pGreenII 0049 pCambia1305.1 pGreenII 0179 pCambia1305.2 pGreenII 0229 pCambia1380 pGreenII 0579 pCambia1381 pHANNIBAL pCambia1381Xa pHELLSGATE pCambia1381Xb pHELLSGATE 12 pCambia1381Xc pHELLSGATE 4 pCambia1381Z pHELLSGATE 8 pCambia1390 pKANNIBAL pCambia1391 pPZP100 pCambia1391Xa pPZP101 pCambia1391Xb pPZP102 pCambia1391Xc pPZP111 pCambia1391Z pPZP112 pCambia2200 pPZP121 pCambia2201 pPZP122 pCambia2300 pPZP200 pCambia2301 pPZP201 pCambia2301-‐101 pPZP202 pCambia3200 pPZP211 pCambia3201 pPZP212 pCambia3300 pPZP221 pCambia3301 pPZP222 pCambia35s-‐ECFP pPZp-‐RCS2-‐Bar pCambia35s-‐EGFP pRI 101-‐AN pCambia35s-‐EYFP pRI 101-‐ONpCambia5105 pRI 201-‐AN pSB1 pRI 201-‐ON pSB11 pRI 909 pSoup pRI 910 pSPYCE(MR) pRI101pTCK303 pSAT1-‐cCFP-‐C Super1300 pSAT1-‐cCFP-‐N pSAT6nCeruleanC(A+) pSAT4-‐nVenus-‐C。

载体结构设计酶切位点和引物原理一、概述随着生物技术的发展,载体结构设计在基因工程研究中扮演着至关重要的角色。
其中,酶切位点和引物的设计原理对于基因克隆、表达及遗传转化等方面均有重要意义。
本文将从酶切位点和引物的原理出发,探讨载体结构设计的相关内容。
二、酶切位点的设计原理1. 基本概念酶切位点是指DNA序列上特定的核苷酸序列,能够被特定的限制性内切酶识别并切割。
通过合理设计酶切位点,可以实现对DNA序列的特异性切割,为后续的基因工程操作提供必要的前提条件。
2. 设计原则(1)选择适当的限制性内切酶酶切位点的选择应基于限制性内切酶的特异性,并考虑到后续的克隆和操作需求。
通常选择常用的限制性内切酶,如EcoRI、BamHI等。
(2)避免酶切位点的相互重叠在设计酶切位点时,应当避免酶切位点之间的相互重叠,以免出现无法正常切割的情况。
还应考虑酶切位点的相对位置,以保证后续的克隆操作能够顺利进行。
(3)考虑DNA序列的完整性在设计酶切位点时,应当考虑到DNA序列的完整性,避免对基因序列造成不必要的破坏。
还需注意酶切位点的分布情况,以减少对DNA序列的干扰。
3. 应用示例以人类胰岛素基因为例,其序列包含多个酶切位点,如EcoRI和BamHI。
通过合理设计酶切位点,可以实现对人类胰岛素基因的特异性切割,为后续的基因工程操作奠定了基础。
三、引物的设计原理1. 基本概念引物是用于PCR扩增、基因克隆等实验操作中的一种寡核苷酸片段。
合理设计的引物能够与目标DNA序列特异性结合,从而实现对目标序列的扩增和克隆。
2. 设计原则(1)选择合适的引物长度引物的长度应控制在15-30个核苷酸之间,过长或过短的引物容易导致PCR扩增效率低下。
(2)避免引物间的相互作用在引物的设计中,需要避免引物之间的相互作用,以免出现不必要的二聚体或引物假扩增。
(3)考虑引物的特异性引物的设计应基于目标DNA序列的特异性,避免引物与非目标序列结合,从而影响后续实验结果的准确性。

常用载体构建说明书步骤:一、基本耗材准备(抗生素、LB液体培养基、LB固体培养基、离心管、枪头、三角瓶)二、制备感受态大肠杆菌三、设计引物四、Pcr扩增五、Pcr产物检测六、Pcr产物回收七、双酶切pcr产物和质粒八、酶切产物回收九、目的基因与载体连接十、转化感受态大肠杆菌十一、单克隆检测十二、测序比对十三、提质粒十四、酶切验证十五、转化感受态农杆菌十六、单克隆检测十七、侵染液配制一、基本耗材准备1、抗生素的制备(抗生素为索来宝公司)常用抗生素Kan(卡那)Amp(氨苄)Rif(利福平)母液浓度:Kan 50mg/ml Amp 100 mg/ml Rif 100 mg/ml工作浓度:Kan 50ng/ul Amp 100 ng/ul Rif 100 ng/ul举例:称取kan固体1g 于注射器中,加入20ml ddH2O,溶解后用过滤器注入灭过菌的离心管中,-20度保存,使用比例1:1000注意:Rif溶解时加入DMSO2、培养基的配制LB液体培养基1000ml 200ml牛肉膏5g 1g蛋白胨10g 2g氯化钠10g 2gLB固体培养基1000ml 200ml牛肉膏蛋白胨氯化钠琼脂粉5g10g10g15g1g2g2g3g120℃高温高压灭菌15-20min,固体培养基冷却后加入相应抗性,加入比例1:1000,然后把培养基倒90cc的培养皿中,一般情况下一个培养皿可倒20ml培养基。
3、离心管、枪头、三角瓶0.2ml、1.5ml、2.0ml离心管各200-500个,50ml离心管2个10ul、200ul、1000ul 枪头各2盒50ml、100ml、250ml 、500ml三角瓶各2个去离子水或双蒸水500ml二、制备感受态大肠杆菌(所用超级感受态细胞制备试剂盒购于上海生工)BT Media 培养基制备:1支BT Media加50 ml蒸馏水配制,放入250ml三角瓶,高压灭菌即可准备工作:将BT Buffer A和BT Buffer B 放冰上遇冷,遇冷低温离心机至4℃1、用超低温冰箱中保存的菌种在LB平板上进行划线,置37℃培养箱中静置培养12-16h待菌落生长到1-2mm大小。
碱基序列和引物序列
碱基序列的研究可以揭示生物体内基因的编码信息,帮助科学家理解基因的功能和调控机制。
通过比较不同生物体的碱基序列,可以揭示它们之间的亲缘关系和进化历史。
此外,碱基序列也被广泛应用于疾病诊断、药物研发和个性化医学领域。
引物序列则是在PCR等实验中用来扩增特定DNA或RNA片段的关键工具。
通过设计特异性的引物序列,科学家可以选择性地扩增感兴趣的基因或DNA片段,从而进行后续的分析和研究。
引物序列的设计需要考虑到目标序列的特异性和合适的引物长度,以确保PCR实验的准确性和灵敏度。
总之,碱基序列和引物序列在生物学研究中扮演着不可或缺的角色,它们的研究和应用为我们深入理解生命的奥秘提供了重要的工具和途径。
蛋白质序列构建质粒的方法蛋白质序列构建质粒是一种重要的分子生物学技术,在生物工程和基因工程领域得到广泛应用。
下面我将详细介绍蛋白质序列构建质粒的方法。
质粒是一种环状双链DNA分子,常见于细菌中,并且具有自主复制能力。
质粒构建是将感兴趣的蛋白质的编码序列插入到质粒DNA中的过程。
这样构建出的质粒可以被细菌或其他真核生物细胞摄取并进行表达,使得该蛋白质在细胞中大量产生。
蛋白质序列构建质粒的方法主要包括以下几个步骤:1. 选择合适的质粒载体:质粒载体是质粒构建的基础。
根据具体需求,可以选择适合的宿主细胞和质粒类型。
常用的质粒载体有pUC、pET等。
2. 扩增目标蛋白质基因:使用PCR或其他扩增方法,将目标蛋白质基因从其来源中扩增出来。
PCR反应需要设计适当的引物,其中一个引物需包含与目标质粒载体相对应的序列。
3. 限制性内切酶切割:购买适当的限制性内切酶,根据目标蛋白质基因和质粒载体的序列设计合适的酶切位点。
将扩增的目标基因和质粒载体经过双酶切,形成互补的粘性末端。
4. 连接反应:将切割后的目标蛋白质基因和质粒载体进行连接反应。
在此步骤中,需要使用DNA连接酶将两者进行连接,并生成一个新的DNA分子。
5. 转化:将连接后的质粒DNA转移到适当的宿主细胞中。
其中,常用的宿主细胞包括大肠杆菌(E. coli)等。
转化方法有化学转化、电转化等。
6. 识别和筛选:将转化后的细菌进行培养和筛选。
在培养基中加入适当的抗生素,只有带有质粒的细菌能够在该培养基中生存,通过抗生素的筛选,可以筛选出带有目标质粒的细菌。
7. 验证和纯化:对得到的带有目标质粒的细菌进行验证和纯化。
验证可以通过PCR或测序等方法进行。
纯化则可以使用柱层析等技术进行。
总之,蛋白质序列构建质粒是一项复杂而重要的分子生物学技术。
通过适当的实验设计和操作,可以高效地构建出带有目标蛋白质编码序列的质粒,为进一步的蛋白质表达和功能研究打下基础。