第二章 配体的合成(43-68)
- 格式:doc
- 大小:932.00 KB
- 文档页数:26

第二单元配合物是如何形成的配合物的命名原则一、配体位次1 . 化学式“原则”在IUPAC1970规定一致,即:(1)“在配位个体中如既有无机配体又有有机配体,则无机配体排列在前,有机配体排列在后。
”例:cis-[PtCl2(Ph3P)2]。
因此,如[Cr(en)2Cl2]Cl,[Co(en)2(NO2)(Cl)]SCN,[Pt(en)CO3]等都不符合(1)。
(2) “无机配体和有机配体中,先列出阴离子,后列出阳离子和中性分子。
”例:K[PtCl3NH3],[Co(N3)(NH3)5]SO4。
因此如[Co(NH3)5Cl]Cl2,[Pt(NH3)2Cl2],[Co (NH3)(CO3)]+,[Co(NH3)3(OH2)Cl2]+,[Co(en)2(NO2)(Cl)]+,[Pt(en)(NH3)(CO3)]等都不符合(2)或5(1)和(2)。
(3) “同类配体的名称,按配位原子元素符号的英文字母顺序排列。
”例:[Co(NH3)5H2O]3+。
据此不能写成[CoH2O(NH3)5]3+。
(4) “同类配体中若配位原子相同,则将含较少原子数的配体排在前面,较多原子数的配体列后。
”例:[PtNO2NH3NH2OH(Py)]Cl。
因此,[Co(NH3)3(NO2)3]不符合(4)。
2 . 命名配体命名的顺序,按“原则”示例可知,与配位个体中中心离(原)子后的配体书写顺序(化学式)完全一致;IUPAC的规则却不同,是按配体的英文名称词头字母(例中有底线者)的英文字母顺序命名,故与化学式的顺序不一致;日本则按阴离子配体、阳离子配体、中性分子配体的顺序命名,与我国的“原则”大体一致。
例:(1)K3[Fe(CN)6]六氰合铁(Ⅲ)酸钾(stock方法),六氰合铁酸(3-)钾(Ewen—Basett方法);potassium hexa cyanoferrate(Ⅲ)或potassium hexa cyanoferrate(3-)(英)以下仅用stock方法。

1,2-二(3-吲哚亚甲胺基)乙烷的合成(希夫碱):第一步:于小烧杯中称取3-吲哚甲醛(11.2g, 0.1mol)置于250ml的圆底烧瓶中,用少量绝对乙醇(5-10ml)涮洗小烧杯,洗液倒入烧瓶中,再加入大约15ml 的绝对乙醇。
滴入1滴浓硫酸,磁子搅拌。
第二步:再于装有适量绝对乙醇(5-10ml)的小烧杯中称取1,2-乙二胺(重蒸)(3.0g, 0.05mol),装于恒压漏斗中,用15ml绝对乙醇涮洗小烧杯,洗液倒入恒压漏斗中,慢慢滴加到圆底烧瓶中,磁子搅拌。
第三步:滴加完毕后,溶液有白色浑浊,室温反应30小时。
其间用TLC做监测。
第四步:将反应后的溶液旋去约一半体积,放入冰箱,3-5℃过夜。
第五步:将产生的沉淀用砂芯抽滤,并用少量冷的无水乙醇洗2-3次,得灰白色固体。
干燥;即为产物。
第六步:将剩余溶液合并,重复四、五步骤。
1,2-二(3-吲哚甲胺基)乙烷的合成:第一步:将1,2-二(3-吲哚亚甲胺基)乙烷(15g, 0.063mol)置于250ml的圆底烧瓶中,冰浴条件下磁子搅拌。
第二步:称取NaBH4(6.0g, 0.16mol;其中NaBH4的量约为希夫碱的量的2.5倍)少量多次加入到烧瓶中,约3-5小时(由于反应剧烈,每次加入量不要太多,又因为硼氢化钠极易受潮,加完后应放到干燥器中保存;每加完硼氢化钠后,应用干燥管塞住烧瓶瓶口),加完后室温磁搅过夜。
(在加入硼氢化钠的过程中,溶液会产生大量气泡,溶液颜色逐渐变深,反应温度至室温后溶液颜色又逐渐变浅为米黄色)。
第三步:约24小时后停止反应,将溶剂旋干,得糊状物。
第四步:加入适量的蒸馏水使得到的固液混合物刚好溶解,用CH2Cl2萃取5次,每次70ml。
合并有机相(CH2Cl2),用无水NaSO4或无水MgSO4干燥3-5小时。
(注意:在萃取过程中,CH2Cl2相一般在下层,但由于水相密度过大,有时在前几次萃取时水相会在下层,要注意分辨)。

配位化学的配位化合物合成配位化学是化学领域中的一个重要分支,研究的是金属离子与配体形成配位化合物的过程。
配位化合物合成的方法多种多样,可以通过配位反应、溶液反应、固体反应等途径来实现。
本文将介绍配位化学的配位化合物合成方法以及一些典型的实例。
一、配位反应法配位反应法是配位化合物合成的常用方法之一。
在配位反应过程中,金属离子与配体之间会发生配位键的生成或断裂,从而形成新的配位化合物。
常用的配位反应方法包括配体置换反应、配体加合反应和配位缩合反应等。
1. 配体置换反应配体置换反应是指用新的配体取代原有配体的过程。
在这个过程中,原有配体会与金属离子发生键的断裂,然后新的配体与金属离子形成新的配位键。
常见的配体置换反应包括水合作用和配体交换反应等。
例如,将氯化镍和亚硝酸钠反应可以得到亚硝酸镍:NiCl2 + 2 NaNO2 → Ni(NO2)2 + 2 NaCl2. 配体加合反应配体加合反应是指两种或多种配体与金属离子同时发生配位键生成的过程。
在这个过程中,多个配体与金属离子形成配位键,生成多核配位化合物。
常见的配体加合反应有配体加合聚合反应和配体加合还原反应等。
例如,二氯化铜和四氯化碳反应可以得到二氯化四氯化碳铜:CuCl2 + CCl4 → CuCl2(CCl4)3. 配位缩合反应配位缩合反应是指由两个或多个配体与金属离子反应生成一个较大的配位化合物的过程。
在这个过程中,两个或多个配体之间发生缩合,形成一个配位聚合物。
常见的配位缩合反应有缩合聚合反应和配位链反应等。
例如,二乙酸铜和巯基乙醇反应可以得到巯基乙醇合铜(II):Cu(O2CCH3)2 + HSCH2CH2OH → HSCH2CH2OOCCH3 +Cu(OOCCH3)2二、溶液反应法溶液反应法是指在溶液中进行配位化合物合成的方法。
在溶液中,金属离子和配体之间发生反应,形成溶液态的配位化合物。
溶液反应法适用于需要在溶液中合成大量配位化合物或需要对反应进行控制的情况。

金属有机化学中的配体设计与合成金属有机化学是一门研究有机配体与金属之间相互作用的学科,其中配体的设计与合成是该领域的重要组成部分。
本文将介绍金属有机化学中配体设计与合成的基本原理和方法,并探讨其在化学催化、药物研发等领域的应用。
一、配体的设计在金属有机化学中,配体的设计是非常关键的一步。
根据金属离子的性质和所需的反应活性,设计合适的配体可以改变金属离子的电子结构和配位环境,从而影响反应的速率和选择性。
1.1 配体的结构特点配体的结构特点直接影响其与金属离子的配位方式和稳定性。
常见的配体结构包括双齿配体、多齿配体和桥联配体等。
双齿配体可以通过两个配位原子与金属离子形成化学键,多齿配体则具有更多的配位原子,可以提供更多的电子密度给金属离子,增强其稳定性和活性。
1.2 配体的电子性质配体的电子性质包括配体中的配位原子和配位基团,可以通过改变它们的电子性质来调控金属离子的反应活性。
例如,引入电子供体基团可以增加金属离子的氧化还原性,而引入电子受体基团可以降低其氧化还原性。
二、配体的合成配体的合成是实现设计理念的关键一步。
合成方法的选择应该考虑到配体的结构和性质,并尽可能地简单高效。
2.1 有机合成方法有机合成方法广泛应用于配体的合成,例如取代反应、格氏反应和偶联反应等。
通过合理选择反应条件和底物,可以合成出具有所需结构和性质的配体。
2.2 过渡金属催化反应过渡金属催化反应在配体的合成中扮演着重要角色。
常用的过渡金属催化反应包括金属催化的碳-碳键形成反应和金属催化的碳-氧键形成反应等。
这些反应可以高效地构建配体的骨架,并引入所需的基团。
三、配体在金属有机化学中的应用配体作为金属有机化学的核心组分,在化学催化和药物研发等领域发挥着重要作用。
3.1 化学催化配体可以改变金属催化剂的电子结构和配位环境,从而调控反应的速率和选择性。
例如,采用手性配体可以实现不对称合成,合成具有特定立体结构的化合物。
3.2 药物研发金属配合物作为药物候选化合物具有广泛的应用前景。


化学实验中的配位化合物合成化学实验中的配位化合物合成是一项常见的实验方法,通过合成可以得到各种不同性质和用途的化合物。
本文将介绍配位化合物合成的基本原理、实验步骤和实验注意事项。
一、配位化合物合成的基本原理配位化合物是由中心金属离子和周围的配体离子或分子通过配位键结合而成的化合物。
合成配位化合物的基本原理是选择适当的中心金属离子和配体,使它们能够形成稳定的配位键。
其中,中心金属离子的选择通常基于其电子构型和化学性质,而配体的选择则考虑到其配位能力和稳定性。
二、配位化合物合成的实验步骤1. 实验准备:根据实验需要,准备所需的中心金属离子和配体,选择适当的溶剂和实验器材。
2. 配位反应:将中心金属离子和配体按一定的比例溶解在溶剂中,通过搅拌、加热或冷却等方法促进反应的进行。
3. 反应产物的分离和纯化:将反应混合物进行过滤、结晶、萃取等操作,分离出目标化合物。
4. 配位化合物的鉴定:通过一系列物理性质和化学性质的测试,确定所合成的化合物的结构和性质。
5. 结果分析:根据实验结果进行数据分析和结论总结,评价合成效果和实验方法的可行性。
三、实验注意事项1. 实验操作要小心谨慎,避免发生意外事故。
根据实验室安全规范,佩戴适当的防护装备。
2. 选择合适的实验条件,如反应温度、pH值等,以保证反应的进行和产物的质量。
3. 注意溶剂的选择和使用,避免对实验结果产生干扰或危害。
4. 实验过程中要注意反应时间和溶解度等因素,避免过度反应或出现沉淀。
5. 在进行结构鉴定时,可以利用光谱分析、元素分析等手段,辅助确定化合物的结构和成分。
6. 在实验结束后,要及时清洗实验器材并做好废弃物处理。
综上所述,化学实验中的配位化合物合成是一项重要的实验技术,在化学研究和应用中起着关键作用。
通过合适的实验步骤和注意事项,能够成功地合成出各种不同性质的配位化合物,并为后续的研究和应用提供有效的材料基础。





第2章配合物的合成第一节利用配体取代反应合成配合物1、水溶液中的取代反应1)用金属盐水溶液直接与配体反应[Cu(H2O)4]SO4+ 4NH3 [Cu(NH3)4]SO4向反应混合物中加入乙醇,就可得到深蓝色的结晶。
不适合与Fe3+、Al3+、Ti4+2)煮沸K3[RhCl6] +3K2C2O4 K3[Rh(C2O4)3] + 6KCl2、非水溶剂中的取代反应使用非水溶剂的原因:A、防止水解(如Fe3+、Al3+、Ti4+);B、使不溶于水的配体可溶解;C、配体的配位能力不及水。
1)[Cr(en)3]Cl3的合成在水中反应时CrCl3.6H2O + en Cr(OH)3↓可在乙醚中,按如下方法合成:en KI AgCl无水Cr2(SO4)3溶液[Cr(en)3]I3 [Cr(en)3]Cl32)[Ni(phen)3]Cl2(phen为邻菲咯啉)NiCl2·6H2O + phen [Ni(phen)3]Cl23)[Ni(EtOH)6](ClO4)2的合成NaClO4无水NiCl2 + EtOH [Ni(EtOH)6]Cl2 [Ni(EtOH)6](ClO4)2在水溶液中:[Ni(EtOH)6]2++ H2O [Ni(H2O)6]2++ EtOH3、固体配合物热分解(固态取代反应)1)[Cu(H2O)4]SO4.H2O = [CuSO4]+5H2O (加热)2)2[Co(H2O)6]Cl2 = Co[CoCl4] +12H2O (加热)变色硅胶的原理(粉红、蓝色)第二节利用氧化还原反应合成配合物1、金属的氧化最好的氧化剂是O2或H2O2,不会引入杂质。
例:[Co(NH3)5Cl]Cl2的合成H2O2 浓HCl CoCl2·6H2O [Co(NH3)5(H2O)]Cl3 [Co(NH3)5Cl]Cl2NH3-NH4Cl 加热2、金属的还原N2H4;NH2OH(产物为N2,不污染产物)[Pt(II)(Ph3P)2Cl2] + Ph3P + N2H4 [Pt(0)(Ph3P)4]第三节利用催化反应制备配合物[Co(NH3)6]Cl3的合成(多相催化)NH3-NH4Cl-H2ORhCl3·3H2O + 2en·2HCl 黄色溶液trans-[Rh(en)2Cl2]NO3↓(金黄色)+ 黄色溶液蒸发溶液cis-[Rh(en)2Cl2]NO3↓(亮黄色)这个例子证明,可利用溶解度差别分离异构体。

配位化合物的合成一、引言配位化合物是指由一个或多个配体与一个中心金属离子或原子通过均衡的配位键形成的化合物。
配位化合物广泛应用于催化剂、药物、材料科学等领域。
本文将介绍配位化合物的合成方法,并探讨其在不同领域的应用。
二、配位化合物的合成方法(一)配体交换法配体交换法是最常见的合成配位化合物的方法之一。
该方法通过将已有配体与待合成配位化合物的前体反应,产生新的配位化合物。
例如,用氯化铜与氰甲烷反应可以合成铜(Ⅰ)氰化物配合物。
(二)配体置换法配体置换法是指将原有配体通过反应与新的配体进行置换,从而合成新的配位化合物。
其中较为常用的方法是通过配体溶剂置换法,即通过在溶剂中加入新的配体,使其与之前的配位化合物反应,生成新的配位化合物。
(三)配体还原法配体还原法是通过将待还原的配体与还原剂反应,从而合成新的配位化合物。
还原剂通常是一种能够提供电子的物质,例如有机金属化合物或铁还原蛋白。
(四)合成气法合成气法是一种通过将配体与合成气(一氧化碳和氢气的混合物)反应,产生金属卡宾或金属氰化物配位化合物的方法。
这种方法具有合成效率高、反应条件温和等优点,被广泛应用于工业生产中。
(五)水热合成法水热合成法是一种利用高温高压条件下水作为溶剂,在反应器中加入金属离子和有机配体,并加热反应得到目标配位化合物的方法。
这种方法具有简单、高度纯化以及特殊形貌调控等优点。
三、配位化合物的应用(一)催化剂配位化合物在催化剂领域中具有广泛应用。
例如,铂金属配位化合物可用作汽车尾气催化剂,用于将有害的一氧化碳转化为二氧化碳和水。
另外,钯金属配位化合物常用于有机合成中,作为强效催化剂。
(二)药物许多药物中也含有配位化合物。
例如,铁配合物在治疗贫血和血液疾病中被广泛应用。
另外,白金配合物是一类广泛用于抗癌药物中的化合物,如顺铂和卡铂。
(三)材料科学配位化合物在材料科学领域中也有重要应用。
例如,铜配合物作为染料敏化太阳能电池中的光吸收剂;金属-有机框架化合物在气体存储、分子分离、催化等领域有广泛应用。




配位体的合成按文献[ 3 ]方法,将邻香草醛与对苯二胺以摩尔比210∶110 ,在无水乙醇中缩合制得橙红色双希夫碱配位体。
配位体熔点为221~222 ℃,与文献值[3 ]一致。
其分子式为C22H20N2O4 ,配合物的合成在加热回流条件下,将含310 mmol 硝酸铀酰(01810 g UO22 + ) 或硝酸钍(01697 g Th4 + )的无水乙醇溶液(20 mL) 慢慢滴加到含310 mmol 配位体的苯溶液中,即有沉淀析出,继续回流3 h (回流速率为2 滴/ s) 。
趁热抽滤,沉淀用热苯洗涤多次,将所得产物室温真空干燥、保存。
1. 2 配体的合成由糠醛与邻苯二胺以2. 0∶1. 0 摩尔比在无水乙醇中进行缩合反应,得到深红色沉淀,再将沉淀用无水乙醇重结晶得双希夫碱配体。
其元素分析结果与按化学式C16 H12N2O2 的计算值相符。
测定值( %) :C ,72. 64 ; H ,4. 52 ;N ,10. 83 ; 计算值( %) :C ,72. 71 ;H ,4. 58 ;N ,10. 60。
1. 3 配合物的合成将含5. 0 mmol Mn (Ac) 2 ·4H2O 的无水乙醇溶液(约20 mL) 慢慢滴加到含5. 0 mmol 糠醛缩邻苯二胺双希夫碱配体的无水乙醇溶液中,室温下继续搅拌反应20 h ,将溶液减压浓缩,放置过夜,有浅灰色沉淀生成。
抽滤,沉淀用无水乙醇重结晶,将产物真空干燥保存。
1. 2 配合物的制备配体的制备: 将0. 015mol 邻苯二胺溶于15mL 无水甲醇中,自恒压漏斗中以30 滴/min 的速度滴入15mL 含0. 036mol 水杨醛的甲醇溶液,搅拌反应,体系变为黄色透明溶液。
滴加完毕后平稳回流4h,冷却至室温后蒸馏,得到橘红色固体,用丙酮重结晶,得橘红色片状晶体H2L。
配体H2L 的合成路线:图1 配体的合成路配合物的制备: 称取0. 001moL 亚胺H2 L 溶于15mL 丙酮中,加入0. 002moL NaOH,搅拌反应至完全溶解,溶液颜色变为橘黄色。
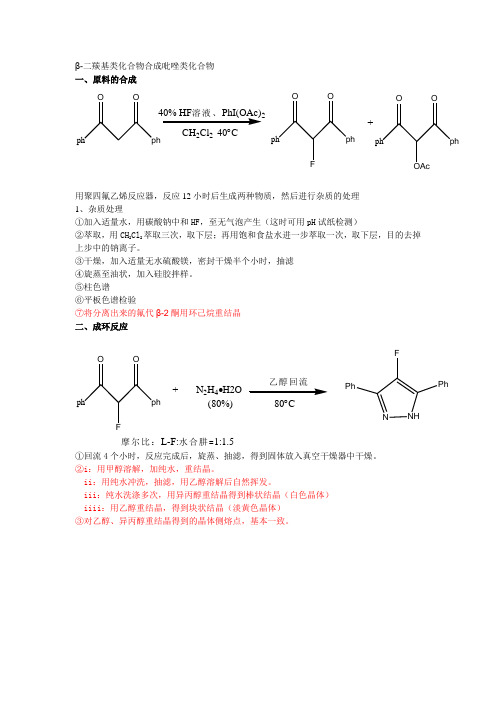
β-二羰基类化合物合成吡唑类化合物 一、原料的合成
ph
ph
O
O
40% HF 溶液、PhI(OAc)2
CH 2Cl 2 40︒C
ph
ph
O
O
F
+
ph
ph
O
O
OAc
用聚四氟乙烯反应器,反应12小时后生成两种物质,然后进行杂质的处理 1、杂质处理
①加入适量水,用碳酸钠中和HF ,至无气泡产生(这时可用pH 试纸检测)
②萃取,用CH 2Cl 2萃取三次,取下层;再用饱和食盐水进一步萃取一次,取下层,目的去掉上步中的钠离子。
③干燥,加入适量无水硫酸镁,密封干燥半个小时,抽滤 ④旋蒸至油状,加入硅胶拌样。
⑤柱色谱
⑥平板色谱检验
⑦将分离出来的氟代β-2酮用环己烷重结晶 二、成环反应
ph
ph
O
O
F
+
N 2H 4∙H2O (80%)
乙醇回流摩尔比:L-F:水合肼=1:1.5
N
NH
Ph
Ph
F
80︒C
①回流4个小时,反应完成后,旋蒸、抽滤,得到固体放入真空干燥器中干燥。
②i :用甲醇溶解,加纯水,重结晶。
ii :用纯水冲洗,抽滤,用乙醇溶解后自然挥发。
iii :纯水洗涤多次,用异丙醇重结晶得到棒状结晶(白色晶体) iiii :用乙醇重结晶,得到块状结晶(淡黄色晶体) ③对乙醇、异丙醇重结晶得到的晶体侧熔点,基本一致。

第二章配体的合成(43-68)第二章配体合成路线的设计及配体的合成43 第二章配体合成路线的设计及配体的合成第一节引言亚磷酰胺配体是一类重要的富电子的有机配体,其过渡金属配合物广泛用于不对称氢化、不对称偶极加成、不对称取代、不对称氢氰化等反应中[116],配体中基团结构的改变、修饰,在催化过程中表现出了不同的行为,某些配体表现出了较高的活性,取得了比较高的转化率和ee值。
但是由于该类配体传统的合成方法操作起来比较麻烦,而且产率普遍不高,因此它们的应用受到了一定的限制。
特别是它们在Suzuki交叉偶联及N -芳基化反应中的应用还未见文献报道。
如何利用简单、易得的原料,通过便捷的方法合成亚磷酰胺配体已成为制约该类配体应用的关键因素。
第二节配体合成路线的设计2.1亚磷酰胺配体的特点与其它富电子的有机配体比较,亚磷酰胺配体这类配体具有如下特点:(1)配体含有磷、氧、氮三类配位原子,可与过渡金属中的钯、铑、铱、铜等配位形成配合物;(2)配体的稳定性较好,特别是固体化合物的稳定性更好一些,便于保存与实验操作,不需要使用手套箱;(3)配体易用于极性及非极性有机溶剂中,对反应介质的要求不高;(4)配体的合成路线简单,通过简单的两步反应即可得到目标化合物;(5)配体合成所用的原料简单、易得,而且价格也比较便宜。
2.2合成亚磷酰胺配体的传统方法亚磷酰胺-过渡金属催化的交叉偶联反应的研究44按照亚磷酰胺配体合成的传统方法,以1,1-联二萘酚为例合成路线如图1所示。
合成路线的第一步是在–78 ℃,在吸酸剂三乙胺存在下,以四氢呋喃作溶剂,1,1-联二萘酚与三氯化磷发生二取代反应,生成中间产物1,1-联二萘酚氯化磷;第二步温度升至-40 ℃,在三乙胺存在下与二级胺发生取代反应得到目标化合物。
反应最终产物的收率大约在80%。
OH OH(i) PCl , Et N THF, -78COOP Cl(ii) R 1R 2NH 3THF, -40 o C OOP NR 1R 2图2-1 亚磷酰胺配体合成的传统方法Scheme 2-1 Traditional route of synthesis of phosphoramidite ligands.2.3合成亚磷酰胺配体的新方法我们对传统的合成方法进行了改进,配体的合成路线如图2所示。


化学中的配体设计与合成在化学中,配体设计和合成是一项非常重要的领域。
配体是有机或无机化合物,它们包含一个或多个可以与金属离子结合的原子或基团。
这些配体在配位化学反应中发挥重要作用,可以通过控制生成的化合物的结构和性能来影响化学反应的效果。
因此,配体设计和合成对于化学研究和应用都具有重要的意义。
一、配体的种类在配位化学中,配体可以分为多种类型,包括有机配体和无机配体。
有机配体是由有机化合物构成的,它们通常含有多个可与金属离子配位的原子或基团。
常见的有机配体有乙二胺 (en)、苯并二氨 (phen)、邻菲罗啉 (dpp) 等。
无机配体是由无机化合物构成的,其中最常用的是水 (H2O) 和氨 (NH3)。
除了有机配体和无机配体之外,还有很多其他类型的配体。
例如,新型的多功能性配体、手性配体等等。
这些新型配体的设计和合成对于新化合物的发现和研究具有至关重要的作用。
二、配体设计的基本原则1.氢键在配体设计中,氢键是一个非常重要的因素,它可以影响配体与金属离子之间的作用强度和发生反应的速度。
例如,一些含有酰胺等氢键供体的配体能够与金属离子形成稳定的配合物。
2.自组装自组装是一种基于非共价相互作用的配体设计方法,它可以通过控制几个化学键之间的作用来控制化学反应的效果。
自组装的优点是可以快速、高效地合成包括配合物、配位聚合物、超分子组装体等有机和无机体系。
3.多功能性分子的设计在配体设计中,多功能性分子的设计非常重要。
这些分子可以同时具有不同的功能 (如荧光性、自组装性、生物活性等),从而为新型化合物的研究提供条件。
三、配体合成方法1.有机合成方法在有机配体的合成过程中,常见的有机合成方法包括缩合反应、亲核取代反应、交叉偶联反应等。
这些反应可以使有机配体的结构变得更加多样化,从而为新型化合物的研究提供条件。
2.无机合成方法在无机配体的合成过程中,最常用的方法包括氧化物沉淀法、热分解法、溶剂热法等。
这些方法可以使无机配体的结构变得更加稳定和多样化。
第二章配体合成路线的设计及配体的合成43 第二章配体合成路线的设计及配体的合成第一节引言亚磷酰胺配体是一类重要的富电子的有机配体,其过渡金属配合物广泛用于不对称氢化、不对称偶极加成、不对称取代、不对称氢氰化等反应中[116],配体中基团结构的改变、修饰,在催化过程中表现出了不同的行为,某些配体表现出了较高的活性,取得了比较高的转化率和ee值。
但是由于该类配体传统的合成方法操作起来比较麻烦,而且产率普遍不高,因此它们的应用受到了一定的限制。
特别是它们在Suzuki交叉偶联及N -芳基化反应中的应用还未见文献报道。
如何利用简单、易得的原料,通过便捷的方法合成亚磷酰胺配体已成为制约该类配体应用的关键因素。
第二节配体合成路线的设计2.1亚磷酰胺配体的特点与其它富电子的有机配体比较,亚磷酰胺配体这类配体具有如下特点:(1)配体含有磷、氧、氮三类配位原子,可与过渡金属中的钯、铑、铱、铜等配位形成配合物;(2)配体的稳定性较好,特别是固体化合物的稳定性更好一些,便于保存与实验操作,不需要使用手套箱;(3)配体易用于极性及非极性有机溶剂中,对反应介质的要求不高;(4)配体的合成路线简单,通过简单的两步反应即可得到目标化合物;(5)配体合成所用的原料简单、易得,而且价格也比较便宜。
2.2合成亚磷酰胺配体的传统方法亚磷酰胺-过渡金属催化的交叉偶联反应的研究44 按照亚磷酰胺配体合成的传统方法,以1,1-联二萘酚为例合成路线如图1所示。
合成路线的第一步是在 –78 ℃,在吸酸剂三乙胺存在下,以四氢呋喃作溶剂,1,1-联二萘酚与三氯化磷发生二取代反应,生成中间产物1,1-联二萘酚氯化磷;第二步温度升至-40 ℃,在三乙胺存在下与二级胺发生取代反应得到目标化合物。
反应最终产物的收率大约在80%。
OH OH (i) PCl , Et NTHF, -78C O O P Cl(ii) R 1R 2NH 3THF, -40 o C O OP NR 1R 2图2-1 亚磷酰胺配体合成的传统方法Scheme 2-1 Traditional route of synthesis of phosphoramidite ligands.2.3合成亚磷酰胺配体的新方法我们对传统的合成方法进行了改进,配体的合成路线如图2所示。
反应的第一步是在0 ℃ - 5 ℃的条件下,无水乙醚作溶剂,过量的二级胺(本身也是吸酸剂)与三氯化磷反应(二级胺:三氯化磷 = 6:1)生成中间产物三胺基磷,第二步用氯化胺作催化剂,以干苯作溶剂,中间体与芳酚在回流下得到目标产物。
R 1R 2NH(i) PCl 3(ii) phenol 1Ar 1-Ar 2:2346Ar 1 = Ar 2:7P O Ar 1Ar 2O NR 1R 2ether, 0 C 2-75NH 4Cl, 80 o C (R 1R 2N)3P a R 1 = R 2 = CH 3b R 1 = R 2= C 2H 5c R 1 = R 2 = Pr i d R 1 = R 2 = CH 2Ph e R 1-R 2 = (CH 2)4f R 1-R 2 = (CH 2)5g R 1-R 2 = (CH 2)2O(CH 2)2 图2-2 亚磷酰胺的配体合成的新方法Scheme 2-2 New route of synthesis of phosphoramidite ligands.第二章配体合成路线的设计及配体的合成45与传统合成方法相比较,新的合成方法具有一下优点:(1)反应温度易于控制,避免了-78 ℃的低温反应;(2)过量的二级胺兼作吸酸剂,与氯化氢生成的盐可以经过与氢氧化钠溶液反应定量的回收,避免了其它吸酸剂的使用;(3)某些固体产物可以通过重结晶的方法而不是较烦琐的柱层析方法得到;(4)即使是通过柱层析方法分离产物,粗产物的处理也比传统的方法容易;(5)该合成方法只要以氩气保护好反应系统,反应产物的收率普遍高于传统方法。
第三节配体合成的实验部分3.1实验仪器与试剂3.1.1实验仪器核磁共振仪:BRUKER DRX 400气相色谱仪:Agilent 4890 D液相色谱仪:Agilent 1100高分辨质谱仪:Mariner 5303熔点仪:YAZAWA熔点仪3.1.2试剂2,2’-联二苯酚AR,二苄胺AR,四氢吡咯AR,吗啡啉AR为Acros 或Aldrich 试剂。
其它试剂均为国产分析纯试剂46亚磷酰胺-过渡金属催化的交叉偶联反应的研究3.2 配体合成的一般过程[117]将96.6 mmol二烷基胺及乙醚的混合液(摩尔比为: 二胺: 乙醚= 1 : 10)在搅拌下冷却至0-5 ℃,并加入16.1 mmol三氯化磷(PCl3)的乙醚溶液(摩尔比为: 三氯化磷: 乙醚= 1 : 30),保持反应温度为0-5 ℃,至白色固状物产生;滴加完毕,升至室温继续搅拌16小时,过滤除去反应产生的固体,蒸去滤液中溶剂,剩余物加入30 mL苯使其溶解,并加入16.1 mmol的二羟基酚(或32.2 mmol的一羟基酚)及0.01 g氯化氨(NH4Cl),加热回流搅拌12小时,以上操作均在氩气保护下进行。
反应毕,蒸发除去溶剂苯,固体产物再以乙醚重结晶,液体产物以石油醚作展开剂,在硅胶柱上进行柱层析,得到目标产物。
按照上述方法合成了2a-2g,3a-3g,4a-4g,5a-5g,6a-6g,7a-7g 共计42种配体。
其中3e,3f,4e,5a,5c,5d,5e,5g,6a,6c,6d,6e,6g,7d,7e未见文献报道。
3.3 过量二级胺的回收将固体二级胺的盐酸盐与氢氧化钠(摩尔比为1 : 1.2)的水溶液混合,在常温下搅拌1小时。
分出有机相,水相用乙酸乙酯萃取3次,合并有机相,用饱和氯化钠水溶液洗涤一次,无水硫酸钠干燥2小时。
常压蒸馏(或减压蒸馏)粗产物即得二级胺(回收率> 99%)。
第四节配体的表征4.1 配体1H NMR、13C NMR及31P NMR谱的测定以CDCl3作溶剂,TMS 作内标,使用Bruker DRX-400 核磁共振仪对合成的配体进行了表征;第二章配体合成路线的设计及配体的合成47测定结果表明:所用配体的1H NMR、13C NMR及31P NMR谱的测定数据与配体的结构相符(见第三章及第四章附录)。
由于受氧原子去屏蔽作用的影响,配体上与氧相连碳的13C NMR向低场移动,δ值在147-154之间,明显高于其它芳环碳的δ值。
刚性比较大的1, 1-联二萘酚、2, 2-联苯二酚及邻苯二酚类磷酸(酰)胺酯配体的31P NMR谱的数值相对比较大,δ在145-156之间;柔性比较大的α-萘酚、β-萘酚及苯酚类磷酸(酰)胺酯配体的31P NMR谱的数值相对比较小,δ在135-145之间4.2配体高分辨质谱(HRMS)的测定以乙氰为溶剂,在内标物存在下,使用飞行时间质谱仪Mariner 5303 (Applied Biosystems, APCI, USA),对合成的42种配体进行了测定。
配体的飞行时间质谱测定结果与配体的分子量相符合(见本章后所附数据及附图)。
值得注意的是,含有二苄氨基的配体在作HRMS谱时有明显分解现象,在HRMS谱上出现两个峰,比较强的峰为分解后的产物二苄胺的峰。
某些含有二苄氨基的配体由于分解比较严重,不能得到HRMS的计算值。
已知化合物的表征数据与参考文献进行了比较[118]。
48亚磷酰胺-过渡金属催化的交叉偶联反应的研究附:配体的高分辨质谱(HRMS)图附图1 HRMS (2a in CH3CN)附图2 HRMS (2b in CH3CN)第二章配体合成路线的设计及配体的合成49附图3 HRMS (2c in CH3CN)附图4 HRMS (2d in CH3CN)50亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图5 HRMS (2e in CH3CN)附图6 HRMS (2f in CH3CN)第二章配体合成路线的设计及配体的合成51附图7 HRMS (2g in CH3CN)附图8 HRMS (3a in CH3CN)52亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图9 HRMS (3b in CH3CN)附图10 HRMS (3c in CH3CN)第二章配体合成路线的设计及配体的合成53附图11 HRMS (3d in CH3CN)附图12 HRMS (3e in CH3CN)54亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图13 HRMS (3f in CH3CN)附图14 HRMS (3g in CH3CN)第二章配体合成路线的设计及配体的合成55附图15 HRMS (4a in CH3CN)附图16 HRMS (4b in CH3CN)56亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图17 HRMS (4c in CH3CN)附图18 HRMS (4d in CH3CN)第二章配体合成路线的设计及配体的合成57附图19 HRMS (4e in CH3CN)附图20 HRMS (4f in CH3CN)58亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图21 HRMS (4g in CH3CN)附图22 HRMS (5a in CH3CN)第二章配体合成路线的设计及配体的合成59附图23 HRMS (5b in CH3CN)附图24 HRMS (5c in CH3CN)60亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图25 HRMS (5d in CH3CN)附图26 HRMS (5e in CH3CN)第二章配体合成路线的设计及配体的合成61附图27 HRMS (5f in CH3CN)附图28 HRMS (5g in CH3CN)62亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图29 HRMS (6a in CH3CN)附图30 HRMS (6b in CH3CN)第二章配体合成路线的设计及配体的合成63附图31 HRMS (6c in CH3CN)附图32 HRMS (6d in CH3CN)64亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图33 HRMS (6e in CH3CN)附图34 HRMS (6f in CH3CN)第二章配体合成路线的设计及配体的合成65附图35 HRMS (6g in CH3CN)附图36 HRMS (7a in CH3CN)66亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图37 HRMS (7b in CH3CN)附图38 HRMS (7c in CH3CN)第二章配体合成路线的设计及配体的合成67附图39 HRMS (7d in CH3CN)附图40 HRMS (7e in CH3CN)68亚磷酰胺-过渡金属催化的交叉偶联反应的研究附图41 HRMS (7f in CH3CN)附图42 HRMS (7g in CH3CN)。