EST-SSSR分子标记的建立
- 格式:doc
- 大小:392.50 KB
- 文档页数:6
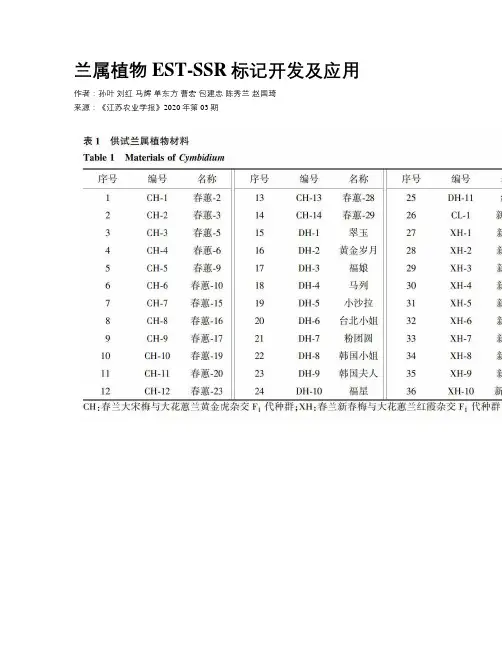
兰属植物EST-SSR标记开发及应用作者:孙叶刘红马辉单东方曹宏包建忠陈秀兰赵国琦来源:《江苏农业学报》2020年第03期摘要:本研究利用蘭属植物杂交种转录组测序数据开发EST-SSR标记,分析标记的多态性及兰属种质的遗传多样性,为兰属植物种质资源的创新和分类研究提供参考。
结果表明,转录组测序分析获得113 780条Unigene,从中共识别到23 709个SSR位点,SSR位点出现频率为20.84%。
从设计的200对引物中筛选出20对具有多态性,并且扩增条带大小与预期相符的EST-SSR标记引物,对48份兰属种质材料进行PCR扩增,平均多态位点数为3.0,共检测到81.00个等位基因,平均每对引物检测到4.05个等位基因。
观测杂合度平均值为0.320 8,期望杂合度平均值为0.507 0;Shannon’s信息指数的变化范围为0.233 8~1.472 4,平均值为0.932 1,多态信息含量为0.110 3~0.662 2。
对4个参试兰属植物种群进行遗传多样性分析,春兰和大花蕙兰的F1杂交种群与大花蕙兰种群亲缘关系较近,与春兰种群的亲缘关系较远。
聚类分析结果表明,遗传相似系数为0.72时,48份种质聚成7类,春兰和大花蕙兰的F1代杂交种群大多聚入第I类,春兰种群聚入第V类,第II类、第III类、第IV类、第VII类均为大花蕙兰种群。
关键词:兰属植物;EST-SSR标记;遗传多样性;亲缘关系中图分类号:S682文献标识码:A文章编号:1000-4440(2020)03-0681-08Development and application of EST-SSR marker in CymbidiumSUN Ye1,2,LIU Hong2,MA Hui2,SHAN Dong-fang2,CAO Hong2,BAO Jian-zhong2,CHEN Xiu-lan2,ZHAO Guo-qi1(1.College of Animal Science & Technology, Yangzhou University, Yangzhou 225009,China;2.Institute of Agricultural Sciences of the Lixiahe District in Jiangsu Province, Yangzhou 225007, China)Abstract: Expressed sequence tag-simple sequence repeat (EST-SSR) markers were developed based on transcriptome sequencing, the polymorphism of markers and the genetic diversity of Cymbidium germplasms were analyzed in order to provide reference for the innovation and classification of resources. The results showed that 113 780 unigenes were obtained from transcriptome sequencing, a total of 23 709 SSR loci were detected in the unigene with the frequency of 20.84%. In addition, 20 pairs of EST-SSR primers with polymorphism were selected from 200 primers, and the size of amplified bands was in accordance with the expectation. Moreover, 48 Cymbidium germplasms were amplified by PCR, and the average number of polymorphic loci was 3.0. A total of 81.00 alleles were observed, with an average of 4.05 alleles per locus. The mean values of observed heterozygosity and expected heterozygosity were 0.320 8 and 0.507 0,respectively. Shannon′s information index ranged from 0.233 8 to 1.472 4, and the average value was 0.932 1. The polymorphic information content(PIC) ranged from 0.110 3 to 0.662 2. The F1 population of Cymbidium georingii and Cymbidium hybridum was close to thepopulation of C. hybridum, and far away the population of C.georingii. Cluster analysis results indicated that 48 germplasms were grouped into seven groups at the genetic similarity coefficient of0.72, the F1 population of C.georingii and C. hybridum was grouped into class I, the population ofC.georingii was grouped into class V, and class II, class III, class IV and class VII were the population of C. hybridum.Key words:Cymbidium;expressed sequence tag-simple sequence repeat (EST-SSR)marker;genetic diversity;genetic relationship兰属植物主要分于中国、日本、韩国、马来群岛、印度西北部、澳大利亚北部和东部等国家和地区,具有很高的观赏价值和经济开发价值。

茶 叶 科 学 2006,26(1):17~23 Journal of Tea Science收稿日期:2005-08-02 修订日期:2005-10-10作者简介:金基强(1983— ),男,河南信阳人,硕士研究生,研究方向为植物诱变遗传与分子改良。
*通讯作者茶树EST-SSR 的信息分析与标记建立金基强1,崔海瑞1*,陈文岳2,卢美贞1,姚艳玲1,忻雅2,龚晓春1(1. 浙江大学原子核农业科学研究所,浙江 杭州 310029;2. 杭州市农业科学院,浙江 杭州 310024)摘要:在1589条茶树EST 中,共发掘出了281个EST-SSR ,分布于246条EST 中,出现频率是17.68%,平均长度为33.06 bp ,平均分布频率是1/2.61 kb 。
在茶树EST-SSR 中,二核苷酸重复是主要的重复类型,出现最多的重复基元类型是AG/CT 重复。
设计了19对SSR 引物,在对引物、dNTP 、MgCl 2的浓度及退火温度等参数进行测试后,建立了合适的PCR 反应体系。
以衍生绝大多数EST-SSR 的龙井43 DNA 为模板,对引物进行了筛选,有16对引物显示扩增,可用率为84.2%;进一步在10个茶树品种中进行多态性测试,显现出多态性的引物占可扩增引物的62.5%。
本文研究结果证明了根据茶树EST 建立SSR 标记是有效、可行的。
关键词:茶树;EST ;SSR 信息;标记建立中图分类号:S571.1; Q523 文献标识码:A 文章编号:1000-369X (2006)01-017-07Data Mining for SSRs in ESTs and Development ofEST-SSR Marker in Tea Plant (Camellia sinensis )JIN Ji-qiang 1, CUI Hai-rui 1*, CHEN Wen-yue 2, LU Mei-zhen 1,YAO Yan-ling 1, XIN Ya 2, GONG Xiao-chun 1(1.Institute of Nuclear and Agricultural Sciences, Zhejiang University, Hangzhou 310029, China; 2. Hangzhou Academy ofAgricultural Sciences, Hangzhou 310024, China)Abstract :Totally 281 SSRs distributed in 246 ESTs were mined out, accounting for 17.68% of 1589 ESTs updated in tea. The average length of tea plant EST-SSRs searched out is 33.06 bp and the average distance of distribution is 1/2.16 kb. The dinucleotide repeat is the dominant type with repeat motif AG/CT being the most common. 19 primer pairs for EST-SSRs were designed. After testing on the annealing temperature and the concentration of primers, dNTP and MgCl 2, a suitable PCR system was established. Under the condition of reaction system developed, the primers designed were screened against genomic DNA of Longjing 43 from which most EST-SSRs were derived, and 16 primer pairs showed the amplification ,accounting for 82.4% of total primers. Then the primers showing amplification were subjected to PCR for DNAs from 10 tea plant cultivars and 10 primer sets showed polymorphisms, accounting for 62.5% of primers available. Results prove that it is an effective and feasible approach to develop SSR markers based on ESTs in tea.Key words :tea plant, ESTs, SSR information, marker development目前尽管已有多种分子标记应用于茶树遗传育种研究[1~4],但其中利用最多的为随机扩增长度多态性DNA(Random amplifiedpolymorphic DNA, RAPD)和限制性片段长度多态性(Amplified fragment length polymorphic, AFLP)等。

园 艺 学 报 2008,35(7):1069-1074Acta Horticulturae Sinica基于EST信息的百合SSR标记的建立杨素丽1,2,明 军13,刘 春1,穆 鼎1,李名扬2(1中国农业科学院蔬菜花卉研究所,北京100081;2西南大学园艺园林学院,重庆400715)摘 要:依据已知的百合EST(exp ressed sequence tags)序列信息,开发新的SSR(si m p le sequence re2 peats)标记,在NCB I的EST数据库1688条EST中检索到98条含有101个SSR的序列,SSR的检出率为5198%,其中三核苷酸重复类型占主导地位,出现频率为2184%。
EST2SSR的重复基元共搜索到47种,其中三核苷酸重复基元类型最为丰富,大约占重复基元类型总数的一半。
利用部分EST2SSR s序列共设计23对SSR引物,以铁炮百合‘Snow Queen’DNA为模板,对引物进行筛选,其中18对引物有扩增产物,占所设计引物总数的78126%;进一步用这些引物在5个杂种系列13个百合品种进行多态性测试,显示多态性的引物占可扩增引物的6617%。
本研究结果证明了基于百合EST信息建立SSR标记是一种有效而又可行的方法。
关键词:百合;EST;SSR;引物设计中图分类号:S68212 文献标识码:A 文章编号:05132353X(2008)0721069206Da t a M i n i n g for S i m ple Sequence Repea tsM arker D evelop m en t i n Expressed Sequence Tags from L ilium L.Y ANG Su2li1,2,M I N G Jun13,L I U Chun1,MU D ing1,and L IM ing2yang2(1Institute of V egetables and F lo w ers,Chinese A cade m y of A gricultural Sciences,B eijing100081,China;2College of Horticulture and L andscape,Southw est U niversity,Chongqing400715,China)Abstract:The gr owing availability of EST sequences fr om L ilium L.p r ovides a potential valuable s ource of ne w SSR markers.I n this study,101SSR2ESTs fr o m1688ESTs in the Nati onal Center for B i otechnol ogy I nf or mati on(NCB I)database,rep resenting5198%of the t otal nu mber of ESTswere identified.Among the m, the trinucleotide repeat is the do m inant type with repeat motifs being the most common,accounting f or2184% of ESTs.Forty2seven kinds of repeat motifs were m ined out fr om all SSR s.T wenty2three SSR p ri m ers were de2 signed t o sequence flanking SSR s,the p ri m er pairs designed were screened against genom ic DNA of‘Snow Queen’fr om which most EST2SSR s were derived,and18p ri m er pairs showed the a mp lificati on,accounting for78126%of t otal p ri m ers.Then the p ri m ers showing a mp lificati on were subjected t o PCR f or DNA fr om13 L ilium L.cultivars of5hybridis m series and12p ri m er pairs showed poly mor phis m s,accounting for6617%of p ri m ers available.Results p r oved that it is an effective and feasible app r oach t o devel op SSR markers based on ESTs in L ilium L.Key words:L ilium L.;EST(exp ressed sequence tags);SSR(si m p le sequence repeats);p ri m er designEST(ex p ressed sequence tags)是一段c DNA5′或3′端的序列,长度一般为150~500bp,大约有1%~5%的EST含有可建立标记的SSR(si m p le sequence repeats)(Kantety et al.,2002)。

园 艺 学 报 2006,33(3):549~554Acta Horticulturae Sinica白菜EST2SSR信息分析与标记的建立忻 雅1,3 崔海瑞13 卢美贞1 姚艳玲1 金基强1 林容杓2 崔水莲2(1浙江大学农业与生物技术学院原子核农业科学研究所,浙江杭州310029;2忠南大学园艺系,大田305764,韩国; 3杭州市农业科学研究院生物研究所,浙江杭州310024)摘 要:数量迅速增加的EST为开发新的SSR标记提供了宝贵的资源。
本研究对4584条白菜EST进行了搜索,共检索出474个SSR,检出率为1013%,包括40种重复基元。
其中二核苷酸和三核苷酸重复单元的EST2SSR占主导地位,二者出现的频率基本相近,占总SSR的近83%;其它重复类型所占比例均不足5%。
G A和G AA是二、三核苷酸中的优势重复类型,分别占二、三核苷酸重复类型的7115%和3715%。
设计了15对EST2SSR引物,在合适的PCR反应体系下,以构建EST的白菜自交系A的DNA为模板,对设计的EST2SSR引物进行筛选,发现15对EST2SSR引物都能扩增出产物。
进一步用这些可扩增的引物对28个白菜品种进行PCR扩增,发现7对引物显示多态性,占引物总数的4617%。
此结果表明,根据EST建立EST2SSR标记是一条简便而又有效的途径。
关键词:白菜;EST;SS R信息;标记中图分类号:S63413 文献标识码:A 文章编号:05132353X(2006)0320549206Da t a M i n i n g for SSRs i n ESTs and EST2SSR M arker D evelopm en t i n Ch i2 nese CabbageXin Ya1,3,Cui Hairui13,Lu Meizhen1,Yao Yanling1,J in J iqiang1,L i m Yongpyo2,and Choi Suryun2(1Institute of N uclear and A gricultural Sciences,College of A griculture and B iotechnology,Zhejiang U niversity,Hangzhou,Zhe2 jiang310029,China;2D epart m ent of Horticulture,Chungnam N ational U niversity,Taejeon3052764,Korea;3B iotechnology R esearch Institute,Hangzhou A cade m y of A gricultural Sciences,Hangzhou,Zhejiang310024,China)Abstract:Exp ressed sequence tags(ESTs),increased rap idly in number recently,are i m portant re2 s ources f or devel opment of ne w SSR markers.I n this study,4584ESTs of Chinese cabbage were screened and 474SSR s including40kinds of repeat motifs were m ined out,accounting for1013%of ESTs.D inucleotide and trinucleotide repeats,with si m ilar frequency and accounting f or83%t ogether in all SSR s,were dom i2 nant,while the frequency f or other repeat type is bel ow5%each.G A and G AA are the most frequent motifs, accounting f o7115%and3715%in dinucleotide and trinucleotide repeats res pectively.15p ri m er pairs f or EST2SSR s were designed.Under a suitable PCR syste m,p ri m ers were screened against genom ic DNA of in2 bred line A fr om which the c DNA library was constructed,and all the15p ri m er pairs showed the a mp lifica2 ti on.Then all the p ri m ers available were subjected t o PCR f or DNA s fr om28Chinese cabbage varieties and7 p ri m er sets sho wed poly mor phis m s,accounting for4617%of t otal p ri m ers tested.Results indicate that it is a si m p le and effective app r oach t o devel op SSR markers based on ESTs.Key words:Chinese cabbage;EST;SSR inf or mati on;Marker devel opment简单重复序列(Si m p le sequence repeat,SSR)也叫微卫星(M icr osatellites)。

SSR分子标记实验操作及其注意事项SSR(Simple Sequence Repeats ) 又称微卫星DNA,是一类由几个(多为1~6个) 碱基组成的基序(motif )串联重复而成的DNA序列,其长度一般较短,它们广泛分布于整个基因组的不同位置上,每个座位上重复单位的数目及重复单位的序列都可能不完全相同,因而造成了每个座位上的多态性。
SSR标记数量丰富,覆盖整个基因组,而且分布均匀,多态性高,呈共显性遗传,重复性好,操作简便,是一种理想的分子标记技术,被广泛应用于构建基因连锁图分子辅助育种品种鉴定遗传资源的保存等方面。
一、试剂配制1.0.2%亲水binding silence(现配现用):1500μL95%乙醇,7.5μL冰醋酸,再加入3μLbinding silence,混匀后使用。
2.0.5%疏水repel silence(购买后可直接使用)3.TBE母液(5×TBE):Tris Base,54g;硼酸,7.5g;EDTA(0.5M pH8.0),20ml.搅拌溶化,定容至1000ml,无需灭菌,室温保存,母液的pH值应保持在8.3左右。
4.Urea-TBE(一块胶板用量):5×TBE,12ml;尿素,27g.加双蒸水溶解后定容至51ml,使用漏斗过滤。
5.40%丙烯酰胺:丙烯酰胺,380g;甲叉双丙烯酰胺,20g.加热至37℃使之溶解,加水定容至1000ml,用醋酸纤维素滤膜(入Nalge滤器,0.45μm孔径)过滤,棕色瓶避光保存于室温。
6.10%APS(过硫酸铵):超纯过硫酸铵,2g;超纯水,18ml.溶解后,4℃保存。
7.TEMED(购买后可直接使用):电泳级的TEMED可购自Bio-Rad,Sigma或其他供应商。
8.Loading buffer:去离子甲酰胺,50ml;0.5MEDTA(pH8.0),1ml;二甲苯腈蓝cyanol,0.125g;溴酚蓝,0.125g.混匀后置于4℃保存。





课程名称: 分子育种学 指导老师: 崔海瑞 成绩: 实验名称: EST-SSR 标记的开发 实验类型: 综合型分工: 谢奕-EST 获取+结果分析+引物设计 王鹏潮-SSR 筛选+SSR 统计+结果分析一、实验目的和要求1、了解SSR 标记的开发策略;2、明确EST-SSR 标记的特点和建立EST-SSR 标记的原理;3、熟悉EST-SSR 标记的过程,掌握EST-SSR 标记的开发技术。
二、实验内容和原理 1、实验内容通过从现有的EST 文库中获取序列,再用软件对其进行SSR 查找与引物设计。
2、实验原理 1)微卫星DNA真核基因组中存在着大量的串联重复序列, 按重复单位的大小, 串联重复可分为卫星(重复序列>70bp )、小卫星(6~70bp )和微卫星DNA (1-6bp )。
微卫星(Microsatellite, MS )是指基因组中以少数几个核苷酸(多数为2~4个)为单位多次串联重复组成的长达几十个核苷酸的序列,又称简单序列重复(Simple Sequence Repeats , SSR )或短串联重复(Short Tandem Repeats , STR )、或简单序列长度多态性(Simple Sequence Length Polymorphism ,SSLP )。
重复数目是可变的,重复序列两侧都有物种特异性的保守序列,所以通过设计引物进行PCR 扩增,就可以检测到不同的DNA 区域重复数目的多态性。
2)SSR 标记的开发策略SSR 包括基因组SSR (genomic SSR 或gSSR )和表达区EST-SSR (genic-SSR 或EST-SSR )。
gSSR 是基于基因组序列开发的,开发起来费时费力,而且因为探针的缘故,种类比较局限。
而EST —SSR 则是存在于表达的基因序列内的SSR ,不包括内含子及非表达的调控区等之中的SSR ,通常三个碱基重复的占多数,与不引起基因翻译过程中移码现象的发生相一致。

火龙果EST—SSR分子标记反应体系的建立与优化作者:杨仕美乔光文晓鹏来源:《山地农业生物学报》2018年第03期摘要:基于火龙果转录组测序序列设计引物,以生物学性状差异明显的11份火龙果种质DNA为材料,采用正交试验设计,对影响火龙果EST- SSR-PCR扩增的2×Taq PCR MasterMix、引物及模板DNA浓度等因素进行了优化,在此基础上对变性聚丙烯酰胺凝胶质量浓度及上样量进行筛选确定。
以期建立适合火龙果的EST-SSR-PCR最佳反应体系。
结果表明,优化后的火龙果EST- SSR-PCR扩增的最佳反应体系为:10 μL混合反应体系含基因组DNA 30 ng、6μL 2×PCR Mix和0.6μL(10 -5mol/L)的EST-SSR引物。
在聚丙烯酰胺凝胶电泳检测中,10%的变性聚丙烯酰胺凝胶质量浓度、2.5μL上样量扩增效果最佳。
该体系的建立可为今后利用EST-SSR标记对火龙果遗传多样性分析、系统发育研究、遗传图谱构建、基因定位和分子标记辅助育种等研究提供基础。
关键词:火龙果;SSR标记;体系优化中图分类号:S667.9文献标识码:A文章编号:1008-0457(2018)03-0014-07 国际DOI编码:10.15958/ki.sdnyswxb.2018.03.003Establishment and Optimization of EST-SSR Marker Methodology in PitayaYANG Shimei1,2,QIAO Guang2,WEN Xiaopeng2*(1.College of Life Science, Guizhou University, Guiyang, Guizhou 550025, China; 2. Institute of Agro-bioengineering/The Key Laboratory of Plant Resources Conservation and Germplasm Innovation in Mountainous Region (Ministry of Education), Guizhou University,Guiyang, Guizhou 550025, China )Abstract:Based on the RNA-Seq of Hylocereus polyrhizuse, EST-SSR markers were investigated. SSR primers were designed and synthesized. Genomic DNA of pitaya germplasms from 11 obviously distinguishable phenotypes were used to carry out PCR. To optimize the experimental system, the effect degree of 2×Taq PCR MasterMix, primers and DNA template concentration was analyzed respectively through orthogonal test. Furthermore, the suitable loading quantity andconcentration for de-naturing polyacrylamide gel electrophoresis were determined. The results showed that the optimal amplification system was 10 μL mixture containing 30 ng DNA template,6μL2×PCR Mix,and 0.6 μL (10-5 mol/L) EST-SSR primer. The optimal concentration for de-naturing polyacrylamide gel electrophoresis was 10%,and the loading quantity was 2.5 μL. Our results indicated that the optimized SSR-PCR system on pitaya germplasms would provide theoretic and technical support for analysis of genetic diversity, establishment of genetic maps, gene localizations and molecular marker assisted breeding.Key words:pitaya; EST-SSR marker; system optimization火龍果(Hylocereus spp.)属仙人掌科(Cactaceae)量天尺属(Hylocereus)和蛇鞭柱属(Seleniereus),是近年来被广泛关注的一种新兴热带、亚热带水果[1]。
EST—SSR分子标记在茶树品种鉴别中的应用摘要:从GenBank数据库中龙井43的EST序列中筛查EST-SSR位点,设计并获得了65对茶树(Camellia sinensis)EST-SSR引物。
以鄂茶1号、龙井43、龙井群体、云抗10号、雪芽100、矮丰的基因组DNA为模板,对所设计的65对EST-SSR引物进行筛选。
结果表明,65对引物均能扩增出清晰条带,有效扩增率为100%;其中有4对引物的扩增产物在6个样本间具有多态性,能用于6个茶树品种的品种鉴别。
关键词:茶树(Camellia sinensis);表达序列标签(EST);简单序列重复(SSR);品种鉴别Abstract:EST-SSR loci were screened from EST sequences of Longjing 43 in the NCBI public database,65 pairs of EST-SSR primers were designed using Primer 5.0 software. The genomic DNA of tea varieties Hubei No.1 tea,Longjing 43,Longjing groups,Yunkang No.10,Xueya 100 and Aifeng were extracted and used as a template to select EST-SSR primers. The results showed that targeted bands of all the 65 pairs of primers were amplified,the amplification rate was 100%. 4 pairs of EST-SSR primers showed polymorphism in the 6 samples,could be used for identification of the 6 tea varieties.Key words:tea(Camellia sinensis);expressed sequence tags(EST);simple sequence repeats(SSR);variety identification茶(Camellia sinensis)是深受广大消费者喜爱的传统农产品,是中国重要的经济作物之一,在国际贸易中也占有较大份额。
蚕豆EST-SSR分子标记的建立及其应用的开题报告一、研究背景蚕豆是一种重要的农业作物,其种子具有高蛋白、低脂肪和多种维生素的特点,被广泛应用于食品、饲料等领域。
然而,蚕豆也存在一些问题,如裂荚、矮性、低产等,严重影响了其生产效益。
因此,开展蚕豆遗传改良研究势在必行。
分子标记技术是分子遗传学的重要手段之一,可以快速、准确地鉴定、分析、筛选物种中的相关基因,在遗传改良中具有重要作用。
EST-SSR分子标记是一种基于EST(表达序列标签)序列的SSR(简单序列重复)标记,由于其具有相对较高的保守性和多态性,因此被广泛应用于植物遗传育种中。
因此,本研究旨在建立蚕豆EST-SSR分子标记库,并应用于蚕豆遗传变异分析和育种研究中,以期为蚕豆遗传改良提供有力的分子遗传学依据。
二、研究内容1. 收集蚕豆EST序列数据,建立EST-SSR分子标记库。
2. 使用PCR技术对EST-SSR标记进行验证和优选,并进行电泳检测,筛选出具有高多态性的EST-SSR标记。
3. 应用优选出的EST-SSR标记对蚕豆不同基因型进行遗传变异分析。
4. 在选育材料中应用EST-SSR标记进行育种研究,探讨标记在蚕豆育种中的应用价值。
三、研究意义1. 通过建立蚕豆EST-SSR分子标记库,可以为蚕豆遗传改良提供重要的分子遗传学依据。
2. 优选出的高多态性EST-SSR标记可以用于蚕豆遗传变异分析和育种研究,有望为蚕豆育种提供新的思路和方法。
3. 本研究对于推进我国蚕豆遗传育种研究,提高蚕豆生产效益,具有一定的实际应用价值。
四、研究方法1. 获取蚕豆EST序列数据并进行处理。
2. 设计EST-SSR引物,进行PCR扩增。
3. 对PCR扩增产物进行电泳检测和分析,筛选出多态性较高的EST-SSR标记。
4. 应用多态性较高的EST-SSR标记对蚕豆进行遗传变异分析和育种研究。
五、预期结果1. 获得一批具有高多态性的蚕豆EST-SSR分子标记。
小麦EST-SSR标记的开发、染色体定位和遗传作图的开题报告一、研究背景小麦是世界上最重要的粮食作物之一,其遗传分析和基因定位是小麦育种和分子遗传学研究的重要部分。
EST-SSR标记是一种基于EST (Expressed sequence tag)的,适用于物种间和品种间的遗传变异分析的分子标记,近年来在小麦分子遗传学研究中得到了广泛应用。
本研究将以小麦为研究对象,利用EST-SSR标记对小麦群体进行遗传变异分析和基因定位,旨在为小麦育种提供分子标记和基因资源。
二、研究目的1.开发小麦EST-SSR标记,为小麦遗传变异分析提供分子标记。
2.利用小麦EST-SSR标记对小麦群体进行遗传变异分析,了解小麦的遗传多样性和种质资源。
3.将小麦EST-SSR标记定位到小麦染色体上,为小麦基因定位提供基础数据。
4.利用小麦EST-SSR标记进行小麦遗传作图,了解小麦基因组结构和性状的遗传规律。
三、研究内容和方法1.开发小麦EST-SSR标记:从公共EST数据库中筛选小麦EST序列,根据序列特征和设计原则,设计EST-SSR引物对。
2.遗传变异分析:对小麦种质进行EST-SSR-PCR扩增和生物信息学分析,获取遗传多样性信息和群体遗传结构。
3.染色体定位:利用小麦EST-SSR标记对小麦染色体上的发掘和映射,确定物理位置和染色体位置。
4.遗传作图:利用小麦EST-SSR标记进行遗传图谱构建,确定关键性状的遗传规律和相关基因。
四、研究意义和预期结果1.本研究将开发小麦EST-SSR标记,并在小麦中大规模应用,为小麦分子育种提供了分子标记和基因资源。
2.遗传变异分析和遗传作图将揭示小麦遗传多样性和性状的遗传规律,为小麦品种改良和精准育种提供科学依据。
3.染色体定位可为小麦基因定位提供基础数据,为小麦基因组研究提供重要资源。
4.预期结果将有助于解决小麦遗传多样性保护、品种鉴定和新品种选育等实际问题,有重要的社会经济价值。
课程名称: 分子育种学 指导老师: 海瑞 成绩: 实验名称: EST-SSR 标记的开发 实验类型: 综合型分工: 奕-EST 获取+结果分析+引物设计 王鹏潮-SSR 筛选+SSR 统计+结果分析一、实验目的和要求1、了解SSR 标记的开发策略;2、明确EST-SSR 标记的特点和建立EST-SSR 标记的原理;3、熟悉EST-SSR 标记的过程,掌握EST-SSR 标记的开发技术。
二、实验容和原理 1、实验容通过从现有的EST 文库中获取序列,再用软件对其进行SSR 查找与引物设计。
2、实验原理 1)微卫星DNA真核基因组中存在着大量的串联重复序列, 按重复单位的大小, 串联重复可分为卫星(重复序列>70bp )、小卫星(6~70bp )和微卫星DNA (1-6bp )。
微卫星(Microsatellite, MS )是指基因组中以少数几个核苷酸(多数为2~4个)为单位多次串联重复组成的长达几十个核苷酸的序列,又称简单序列重复(Simple Sequence Repeats , SSR )或短串联重复(Short Tandem Repeats , STR )、或简单序列长度多态性(Simple Sequence Length Polymorphism ,SSLP )。
重复数目是可变的,重复序列两侧都有物种特异性的保守序列,所以通过设计引物进行PCR 扩增,就可以检测到不同的DNA 区域重复数目的多态性。
2)SSR 标记的开发策略SSR 包括基因组SSR (genomic SSR 或gSSR )和表达区EST-SSR (genic-SSR 或EST-SSR )。
gSSR 是基于基因组序列开发的,开发起来费时费力,而且因为探针的缘故,种类比较局限。
而EST —SSR 则是存在于表达的基因序列的SSR ,不包括含子及非表达的调控区等之中的SSR ,通常三个碱基重复的占多数,与不引起基因翻译过程中移码现象的发生相一致。
建立SSR 标记的前提是必需知道重复序列两列的DNA 序列。
与其他标记相比,SSR 标记具有如下特点: (1)数量较为丰富,覆盖整个染色体组; (2)具有多等位基因特性,信息含量高; (3)以孟德尔方式遗传,呈共显性;(4)易于利用PCR 技术分析,对DNA 数量和纯度要求不高,结果重复性好; (5)每个位点由引物序列决定,便于交换。
近年来,EST-SSR 标记的开发引起关注。
数量迅速增加的ESTs 为开发新的SSR 标记提供了宝贵的资源,各种植物中约有5-10%的EST 含有可用于建立标记的SSR 。
建立EST-SSR 标记要经济得多,而且EST-SSR 标记来源于DNA 的转录区域,比gSSR 标记具有更高的通用性,此外,标记-信息量高。
开发EST-SSR原理基于生物信息学手段的可开发SSR的方法。
在数据库中已存在大量的可免费下载EST序列数据并剔除冗余EST序列,再通过SSR搜索软件获得SSR位点信息,然后根据SSR位点两侧的核酸序列信息的统计与分析,利用软件设计相应的SSR 引物,进而开发SSR标记,最后进行PCR扩增及扩增产物电泳检测。
3)不同的SSR标记开发方法比较传统方法开发SSR:传统的SSR 分子标记开发方法简单、易掌握。
这种方法在许多作物的SSR 标记开发中被广泛应用,现已获得的基因组SSR 标记中,四分之三以上的是利用传统开发方法获得的。
但必须对每个克隆进行筛选鉴定,工作量大,需花费大量人力,财力,而且效率较低,植物中已报道的阳性克隆比率约为2 %~3 % ,成功获得引物的机率则更低。
图1 传统SSR开发方法流程图通过富集策略开发SSR标记:为简化技术环节,提高效率,降低开发成本,通过研究建立了多种采取富积策略的SSR 标记开发方法。
基于RAPD 的开发策略:将RAPD 随机引物与SSR连接,作为PCR扩增引物或者将RAPD扩增产物条带用SSR探针进行Southern杂交,可以有效富集重复序列。
虽然目前尚不清楚RAPD有利于SSR 富集的机理,但这一发现大大激发了人们探求富集SSR方法的兴趣。
PIMA ( PCR Isolation of Microsatellite Arrays)方法:先用RAPD引物从基因组中获得随机扩增片段,扩增片段经载体克隆后,用特异SSR及载体引物对克隆阵列进行PCR检测筛选含SSR克隆,对阳性克隆测序。
以上两种利用RAPD 富集SSR的报道,避免了基因组文库的构建,有利于节省时间和人力、物力,但基于此方法进行大量SSR 分离的成功报道并不多。
4)PCR引物设计原则引物长度:15-30bp,常用为20bp左右。
太短会降低退火温度影响引物与模板配对,从而使非特异性增高;太长则比较浪费,且难以合成。
引物扩增跨度:1kb之是理想的扩增跨度,2kb左右是有效的扩增跨度,而超过3kb 就无法得到有效的扩增. 特定条件下可扩增长至10kb的片段。
引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。
A TGC最好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列。
避免引物部出现二级结构,避免两条引物间互补,特别是3‘端的互补,否则会形成引物二聚体,产生非特异的扩增条带。
引物3’端的碱基,特别是最末及倒数第二个碱基,应严格要求配对,以避免因末端碱基不配对而导致PCR失败。
引物中有或能加上合适的酶切位点,被扩增的靶序列最好有适宜的酶切位点,这对酶切分析或分子克隆很有好处。
引物5'端对扩增特异性影响不大,可在引物设计时加上限制酶位点、核糖体结合位点、起始密码子、缺失或插入突变位点以及标记生物素、荧光素、地高辛等。
通常应在5'端限制酶位点外再加1-2个保护碱基。
引物的特异性:引物应与核酸序列数据库的其它序列无明显同源性所扩增产物本身无稳定的二级结构,以免产生非特异性扩增,影响产量。
引物量:每条引物的浓度0.1~1umol或10-100pmol,以最低引物量产生所需要的结果为好,引物浓度偏高会引起错配和非特异性扩增,且可增加引物之间形成二聚体的机会。
三、主要仪器设备计算机四、操作方法与实验步骤1、EST序列的获取可从数据库中直接下载,主要的数据库有:美国国立生物技术信息中心GenBank/NCBI ( National Center for Biotechnology Information ):;目前有72098808条EST序列,其中植物EST序列24158363条,涉及到895个物种。
其中单子叶植物:7263423条:玉米-2019114;水稻-1333022;小麦-11116126。
欧洲生物信息研究所European Bioinformatics Institute (EBI):/index.html ,包含European Nucleotide Archive;日本DNA数据库DDBJ(DNA Data Bank of Japan):.ddbj.nig.ac.jp/;先以从NCBI下载EST序列为例:首先<EST>下直接填写植物名称后按search,再在<Taxonomy>下搜索栏直接填写植物种属名称后按search,出现以下界面时选EST并点击植物名后,按上述方法下载即可。
2、去冗余(EST序列前处理,在本次实验中由于软件收费略去)采用EST分析软件前处理和剔除冗余EST:直接获取的EST中包含一些低质量片段(<100 bp),少量载体序列及末端存在polyA/T“尾巴”的序列,在开发标记之前应去除这些“噪音”——去冗余。
这些处理可通过一些软件来进行:cross-match(/)或EST-trimmer (.pgrc.ipk-gathersleben.de/misa/download/est-trimmer.pl),去除“尾巴”和屏蔽载体序列。
可用生物信息学软件去冗余:如Cluster W、CD-HIT [/cd-hi/](若只考虑建立标记而不求统计相关参数,也可搜索后聚类,剔除冗余的SSR)。
3、SSR的搜寻可利用以下一些软件在线搜索SSR:(1)RepeatMasker(/cgi-bin/WEBRepeatMasker)(2)SSRIT(/db/searches/ssrtool):①选择搜索参数:最大值;②粘贴序列(<100Kb);③获得结果和统计相关参数。
(3)Sputnik (bri.fr/outils/Pise/spumik.html)也可利用MISA、DNAman、ssrhunter或ssrfinder等进行本地化搜索SSR,注意制定搜索SSR的标准。
4、设计SSR引物利用软件设计相应的SSR引物,常用引物设计软件有primerpremier 5.0和oligo 6.0。
也可以用在线设计软件,如primer3.0 (frodo.wi.mitedu/cgi-bin/primer3/primer3_. Cgi)进行引物设计。
5、建立标记(略去)PCR实验:温度、浓度、[混合模板]扩增多态性及其检测:不同材料、聚丙烯酰胺凝胶电泳、银染五、实验数据记录和处理选取NCBI的EST数据库中小麦Triticum aestivum中的前1000条进行SSRIT分析,后选取5条用primeprime5.0进行引物设计,得到如下结果。
表1 1000条小麦EST中SSR的出现频率重复类型基元种数SSR数目占全部SSR比例(%)出现频率(%)单核苷酸2510.60.5二核苷酸41123.4 1.1三核苷酸182961.7 2.9四核苷酸22 4.30.2总计2647100 4.7表2 1000条小麦EST二核苷酸和三核苷酸重复基元分析重复基元数量发生频率(%)所占比例(%)单核苷酸c/g30.360.0 t20.240.0二核苷酸tc/ga80.872.7 tg10.19.1 at10.19.1 ag 1 0.1 9.1三核苷酸ggc/gcc 6 0.6 20.7 cgg/ccg 4 0.4 13.8 gcg 3 0.3 10.3 tcg/cga 2 0.2 6.9 ggt/acc 2 0.2 6.9 gca 2 0.2 6.9 gag 2 0.2 6.9 ctg 2 0.2 6.9 tcc 1 0.1 3.4 gac 1 0.1 3.4 cac 1 0.1 3.4 ata 1 0.1 3.4 agc 1 0.1 3.4 aag 1 0.1 3.4表3 1000条小麦EST中SSR的重复长度和性质重复基元长度(bp)重复性质变化围平均长度完全重复不完全重复总计二核苷酸14-2414.411011 三核苷酸15-14122.329029 四核苷酸202020 2 合计14-1411847047表4 对小麦EST-SSR设计的引物(其中5对)引物编号重复基元预期产物bp Tm℃GC% 序列(5’→3’)gi|32457 5488 tg(12)343 56.1 50 GGAGTATGTATGTCGAGCCAGT54.2 57.9 CCTCTTGACCTACCCGTTCgi|38374 8333 gcg(5)346 60.4 66.7 TAGAACCACCCGCCCGTC58 57.9 GTGCTGCTGTGCTGATGGAgi|38374 8249 gcc(5)282 58.5 64.7 CTGTCGTGGCGGTCCAA56.4 57.9 CTTCAGCCAGCAGCAAGTCgi|38374 8634 gag(6)276 51.5 50 GATGGGTCCTTGGATTTC49.2 60 CGCCGTGATAAACCCgi|38374 8369 ga(6)185 56.6 39.1 TGAGAAATTCAGGGTAAACAGTG57.2 61.1 CCACCGCCGACTACTGAA六、实验结果与分析从结果中可以得知,小麦EST序列中的SSR种类较为丰富,达到26种之多,主要以三核苷酸重复为主(推测三核苷酸重复类型居多的可能是因为EST编码区序列以三核苷酸重复类型为主,而遗传密码子均为三联体核苷酸类型),其中g/c组合成的SSR所占比例较高,但SSR整体的出现频率并不高,不足5%,平均长度为18bp。