第三章 红外吸收光谱
- 格式:ppt
- 大小:18.35 MB
- 文档页数:66






第三章红外吸收光谱(Infrared Absorption Spectroscopy)3.1 概述红外光谱又称为分子振动光谱或分子振转光谱1、特点:特征性强,适应范围广。
有机、无机、高分子化合物;固态、液态、气态样品都可以进行测定红外分为三个区域,近红外区(0.76μm~2.5μm,12820~4000cm-1)、中红外区(2.5μm~25μm, 4000~400cm-1)和远红外区(25μm~1000μm, 400~33cm-1)。
绝大多数有机化合物的基团震动频率处于中红外区。
2、表示方法:红外光谱多用透光率T%为纵坐标,表示吸收强度,以波数ζ(cm-1)为横坐标,表示吸收峰的位置。
也有用吸光度A为纵坐标,出反峰。
波数是频率的一种表示方法(每厘米长的光波中的波的数目)ζ(cm-1)=波数(cm-1)=1/波长(λcm)=104/波长(μm)=1/λ(cm);ζ·λ=1cm 3、红外光谱产生的基本条件1)E红外光=△E分子振动或υ红外光=υ分子振动2)分子振动时其偶极矩(μ)必须发生变化,即△μ≠0,μ=δr3.2 红外光谱与分子结构的关系3.2.1分子的振动形式*基频:分为两大类:伸缩振动和弯曲(变型)振动。
用υs表示对称伸缩,用υas 表示不对称伸缩,δ表示面内弯曲振动,γ表示面外弯曲振动。
以亚甲基为例:此外,还有一些其它的振动吸收峰存在:*倍频:由振动能级基态跃迁到第二,第三激发态时所产生的,不是整数倍。
*组合频:一种频率红外光,同时被两个振动所吸收。
倍频和组合频统称为泛频,在谱图中均显示为弱峰。
*振动偶合:当相同的两个基团相邻,且振动频率相近时,会发生振动偶合裂分,成为两个峰。
*费米共振:基频与泛频之间发生的振动偶合。
当泛频峰与某基峰相近时,发生相互作用,使原来很弱的泛频吸收峰增强。
图3-12费米共振和倍频。
3.2.2 红外光谱的分区(1)基团结构与振动频率的关系表3-1 基团振动频率与化学键力常数的关系(化学键种类)基团化学键力常数(K/N·cm-1) 键长(Â)振动频率(cm-1)C—C(三键)12~18 1.27 2262~2100C—C(双键)8~12 1.40 1600~1800C—C(单键)4~6 1.54 1000~1300(弱)表3—2基团振动频率与原子折合质量的关系(原子种类)基团折合质量键长(Â)振动频率cm-1C—H 0.9 1.12 2800~3100C—C 6 1.54 约1000C—Cl 7.3 1.77 约625C—I 8.9 2.31 约5000—H N—H 0.971.0336003300-3500(2)基团频率区的划分(表3-3)前三个区域(氢键区、叁键及累积双键区、双键区,即4000——1500 cm-1)称为特征频率区,小于1500 cm-1的区域称为指纹区(单键区,有些文献中以1350 cm-1作为二者的界限)。
第三章红外吸收光谱分析3.1概述3.1.1红外吸收光谱的基本原理红外吸收光谱法又称为分子振动转动光谱,属于分子光谱的范畴,是有机物结构分析的重要方法之一。
当一定频率的红外光照射分子时,若分子中某个基团的振动频率和红外辐射的频率一致,两者产生共振,光的能量通过分子偶极矩的变化传递给分子,该基团就吸收了这个频率的红外光,产生振动能级跃迁;如果红外辐射的频率和分子中各基团的振动能级不一致,该频率的红外光将不被吸收。
如果用频率连续变化的红外光照射某试样,分子将吸收某些频率的辐射,引起对应区域辐射强度的减弱,用仪器以吸收曲线的形式记录下来,就得到该试样的红外吸收光谱,稀溶液谱带的吸光度遵守Lambert-Beer定律。
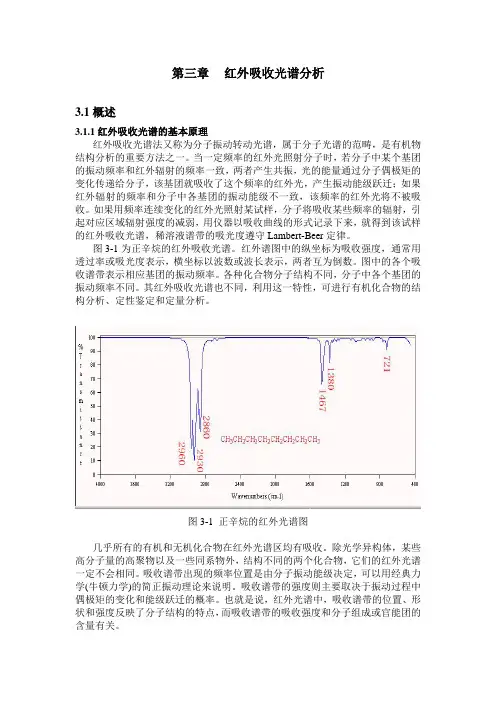
图3-1为正辛烷的红外吸收光谱。
红外谱图中的纵坐标为吸收强度,通常用透过率或吸光度表示,横坐标以波数或波长表示,两者互为倒数。
图中的各个吸收谱带表示相应基团的振动频率。
各种化合物分子结构不同,分子中各个基团的振动频率不同。
其红外吸收光谱也不同,利用这一特性,可进行有机化合物的结构分析、定性鉴定和定量分析。
图3-1 正辛烷的红外光谱图几乎所有的有机和无机化合物在红外光谱区均有吸收。
除光学异构体,某些高分子量的高聚物以及一些同系物外,结构不同的两个化合物,它们的红外光谱一定不会相同。
吸收谱带出现的频率位置是由分子振动能级决定,可以用经典力学(牛顿力学)的简正振动理论来说明。
吸收谱带的强度则主要取决于振动过程中偶极矩的变化和能级跃迁的概率。
也就是说,红外光谱中,吸收谱带的位置、形状和强度反映了分子结构的特点,而吸收谱带的吸收强度和分子组成或官能团的含量有关。
因此,红外吸收光谱在化学领域中的应用,大体上可分为两个方面,即分子结构的基础研究和用于化学组成的分析。
首先,红外光谱可以研究分子的结构和化学键。
利用红外光谱法测定分子的键长和键角,以此推断出分子的立体构型;利用红外光谱法测定分子的力常数和分子对称性等,根据所得的力常数就可以知道化学键的强弱;由简正频率来计算热力学函数等等。


第三章红外吸收光谱法§ 3.1概述分子中基团的振动和转动能级跃迁产生:振-转光谱一、红外光区的划分红外光谱在可见光区和微波光区之间,波长范围约为0.75 ~ 1000 m,根据仪器技术和应用不同,习惯上又将红外光区分为三个区:近红外光区(0.75 ~ 2.5 m ),中红外光区(2.5~ 25 m ),远红外光区(25 ~ 1000 呵)。
近红外光区的吸收带(0.75 ~ 2.5皿)主要是由低能电子跃迁、含氢原子团(如O-H、N-H、C-H )伸缩振动的倍频吸收产生。
该区的光谱可用来研究稀土和其它过渡金属离子的化合物,并适用于水、醇、某些高分子化合物以及含氢原子团化合物的定量分析中红外光区吸收带(2.5 ~ 25 m )是绝大多数有机化合物和无机离子的基频吸收带(由基态振动能级(..=0)跃迁至第一振动激发态(..=1 )时,所产生的吸收峰称为基频峰)。
由于基频振动是红外光谱中吸收最强的振动,所以该区最适于进行红外光谱的定性和定量分析。
同时,由于中红外光谱仪最为成熟、简单,而且目前已积累了该区大量的数据资料,因此它是应用极为广泛的光谱区。
通常,中红外光谱法又简称为红外光谱法。
远红外光区吸收带(25 ~ 1000 m )是由气体分子中的纯转动跃迁、振动-转动跃迁、液体和固体中重原子的伸缩振动、某些变角振动、骨架振动以及晶体中的晶格振动所引起的。
由于低频骨架振动能灵敏地反映出结构变化,所以对异构体的研究特别方便。
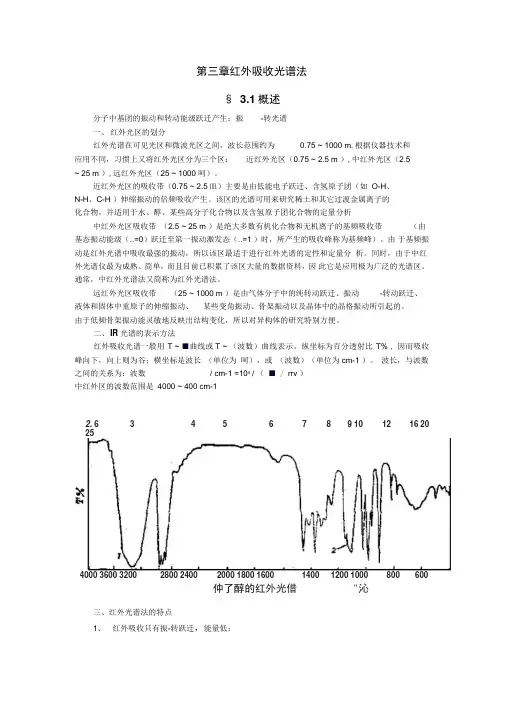
二、IR光谱的表示方法红外吸收光谱一般用T ~ ■曲线或T ~ (波数)曲线表示。
纵坐标为百分透射比T% , 因而吸收峰向下,向上则为谷;横坐标是波长(单位为呵),或(波数)(单位为cm-1 )。
波长,与波数之间的关系为:波数/ cm-1 =104/ (■ / rrv )中红外区的波数范围是4000 ~ 400 cm-12. 6 3 4 5 6 7 8 9 10 12 16 20254000 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600仲了醇的红外光借"沁三、红外光谱法的特点1、红外吸收只有振-转跃迁,能量低;2、应用范围广,除单原子分子及单核分子外,几乎所有的有机物均有红外吸收;3、分子结构更为精细的表征:通过波谱的波数位置、波峰数目及强度确定分子基团和分子结构;4、气体、液体、固体样品都可测定;5、具有用量少;分析速度快;不破坏样品。