中间体检验单
- 格式:xls
- 大小:17.00 KB
- 文档页数:1


阿魏酸哌嗪(保肾康)片中间体检验操作规程一、范围:本标准规定了阿魏哌嗪片(保肾康)片中间体检验方法和操作要求;适用于本公司阿魏哌嗪片(保肾康)片中间体的质量检测。
二、引用标准:企业内控标准三、质量指标:四、试剂1、纯化水五、对照品、阿魏酸哌嗪六、仪器与用具1、紫外分光光度仪2、容量瓶(100ml,250ml)3、移液管(5ml)4、三角瓶(150ml)5、滤纸6、研钵7、电热恒温干燥箱七、操作步骤:1、性状:本品为类白色颗粒。
2、外观:目测无杂点、粗细颗粒均匀。
外事审批程序第八条出国(境)和邀请来华工作政策性强,总公司各部门、专业公司和各子公司要严格按照规定的程序办理各项手续。
第九条出国(境)工作程序为:呈报出国(境)任务申请报告;下达任务批件;办理政审手续并下达政审批件;办理护照和签证。
(一)出国(境)任务申请报告由有出访任务并负责组团的部门和公司自行撰拟上报。
内容包括:出访国家、出国任务、旅行路线、往返时间、出访人员、费用支出等,出访人员不得擅自绕道、增访国家或地区,如确属业务必须,需事先请示总公司外事办公室报总经理批准。
(二)出国(境)任务报告由外事办公室审核(必要时同有关部门会签),报总公司领导审批后向有关单位下达出国(境)任务通知书。
如出访团组中有外单位人员,由外事办公室按授权范围和规定负责向该单位下达出国(境)任务通知书。
(三)出国(境)人员单位应对拟派出人员的政治立场、业务素质、思想作风、近期表现。
身体状况及家庭情况等进行如实填写,实事求是地写出审查意见后报人事本部进行政审。
(四)凡人事档案关系不在总公司的人员,须由所在单位办理政审批件和出国护照;子公司人员、借调人员和聘用的离退休人员,须由原所在单位办理政审批件和出国护照。
(五)出国(境)人员政审(复审)合格后由人事本部办理政审批件。
(六)出国(境)人员持政审批件交外事办公室,由外事办公室统一办理护照和签证手续。
第十条邀请来访工作程序(一)总公司有关部门、专业公司、子公司和控股公司提出邀请来访报告(内容应包括来访者的国家或地区、来访人姓名、身份、人数、来访目的、日程安排、接待方案等),报总公司领导批准后,由总公司外事办公室下达邀请来访任务批件。

药学研制现场核查常见缺陷药学研制现场核查(以下简称研制现场核查)的目的主要是通过对药学研制情况(包括处方与工艺研究、样品试制、质量控制研究、稳定性研究等)的原始资料进行数据可靠性的核实和/或实地确证,核实相关申报资料的真实性、一致性。
本文对药学研制现场核查常见缺陷进行列举,供业内同仁参考。
一、质量管理开展药物研究,应当建立与研究内容相适应的组织机构和质量管理体系,应当具有与药物研究内容相适应的人员、设施、设备、仪器等,制订相应的管理制度或标准操作规程并遵照实施。
常见缺陷示例:1、企业未能配备足够数量并具有适当资质的质量控制管理和操作人员;未对操作人员进行所申报品种的相关培训,或培训日期晚于试验日期。
2、样品试制及检验所用设备无标准操作规程、无使用和维修记录,对于国家强制检定器具目录内的设备未进行未定期进行校验/检定。
称样量记录与天平的精度不符。
3、委托研究存真实性问题,如委托单位的成立日期晚于研究合同签署日,委托试验时间与样品试制完成时间有矛盾。
二、处方和工艺处方和工艺研究过程应当科学完整、合理设计,相关研究记录应当真实完整,与申报资料一致。
常见缺陷示例:1、申报资料中的部分处方工艺研究内容在原始记录中未见记载。
2、申报资料与原始记录不一致,如精储工序关键工艺参数为“收集***流量为5L/h”,而批生产记录中时间点***和***采出液流量分别为3L∕h和2L/h;批生产记录中显示成品有充氮气过程,申报资料中未体现该步骤。
3、实际生产工艺与记录不一致,如在配药后灌装前增加IOUm聚丙烯滤芯过滤去除可见异物,但在工艺规程、批生产记录、申报资料中均未体现增加的过滤工序及滤芯相关信息,亦未查见滤芯与药液的相容性研究资料。
三、样品试制研制样品试制记录,特别是关键批次样品的试制记录应当完整保存。
关犍批次样品的处方和生产工艺、过程控制、试制场地和生产线、使用的主要生产设备型号、技术参数及原始记录等应当与申报资料一致。
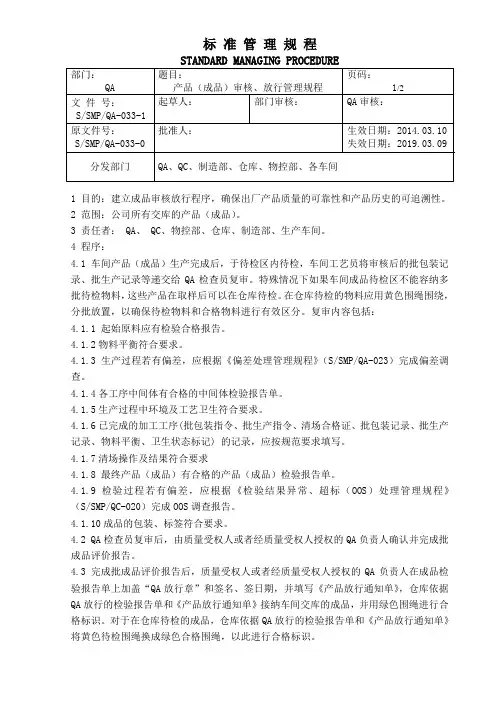
1 目的:建立成品审核放行程序,确保出厂产品质量的可靠性和产品历史的可追溯性。
2 范围:公司所有交库的产品(成品)。
3 责任者: QA、 QC、物控部、仓库、制造部、生产车间。
4 程序:4.1 车间产品(成品)生产完成后,于待检区内待检,车间工艺员将审核后的批包装记录、批生产记录等递交给QA检查员复审。
特殊情况下如果车间成品待检区不能容纳多批待检物料,这些产品在取样后可以在仓库待检。
在仓库待检的物料应用黄色围绳围绕,分批放置,以确保待检物料和合格物料进行有效区分。
复审内容包括:4.1.1 起始原料应有检验合格报告。
4.1.2物料平衡符合要求。
4.1.3 生产过程若有偏差,应根据《偏差处理管理规程》(S/SMP/QA-023)完成偏差调查。
4.1.4各工序中间体有合格的中间体检验报告单。
4.1.5生产过程中环境及工艺卫生符合要求。
4.1.6已完成的加工工序(批包装指令、批生产指令、清场合格证、批包装记录、批生产记录、物料平衡、卫生状态标记) 的记录,应按规范要求填写。
4.1.7清场操作及结果符合要求4.1.8 最终产品(成品)有合格的产品(成品)检验报告单。
4.1.9 检验过程若有偏差,应根据《检验结果异常、超标(OOS)处理管理规程》(S/SMP/QC-020)完成OOS调查报告。
4.1.10成品的包装、标签符合要求。
4.2 QA检查员复审后,由质量受权人或者经质量受权人授权的QA负责人确认并完成批成品评价报告。
4.3 完成批成品评价报告后,质量受权人或者经质量受权人授权的QA负责人在成品检验报告单上加盖“QA放行章”和签名、签日期,并填写《产品放行通知单》,仓库依据QA放行的检验报告单和《产品放行通知单》接纳车间交库的成品,并用绿色围绳进行合格标识。
对于在仓库待检的成品,仓库依据QA放行的检验报告单和《产品放行通知单》将黄色待检围绳换成绿色合格围绳,以此进行合格标识。
4.4同意放行的批次,其生产记录,由QA检查员整理归档,放行交库的产品(成品)方可进行销售。