药物临床试验简介
- 格式:ppt
- 大小:1.63 MB
- 文档页数:26


临床试验、药物试验、医疗器械临床试验管理制度(试行)
2009-11-12 来源:成都市第五人民医院
临床试验、药物试验、医疗器械临床试验管理制度(试行)
一、根据卫生部、国家药品食品监督管理局相关管理规定制定本管理制度。
二、临床试验、药品试验及医疗器械临床试验的前提是负责试验部门必须符合正当的道德原则、符合《赫尔辛基宣言》、符合《人体生物医学研究国际道德指南》、符合《医疗器械临床试验规定》的原则。
三、审批程序
(一)临床药物、医疗器械临床试验申办者在进入临床试验之前,须向科教科提出委托研究的委托函,出具国家食品药品监督管理局的批件、药械部门的检测报告、临床试验方案、病例报告表、知情同意书、研究者手册等,由科教科出具是否接受委托的答复函,并交伦理委员会讨论。
(二)医院伦理委员会到会人数须大于等于总人数的2/3。经伦理委员会讨论同意并在医务科备案后方可进行临床药物、医疗器械临床试验。
四、受试者参加临床试验之前,须保证其在自愿、知情的情况下签署知情同意书,不得以给予报酬等条件进行诱导,或对不愿参加的受试者进行指责或歧视等,受试者的权益和个人隐私权应得到充分保护。
五、 临床试验用药品、医疗器械的生产制造、处理贮存均应符合国家规定,并与试验方案中的规定一致。药械应由申办方准备和提供。进行临床试验前,申办者必须根据各期临床试验提供与临床试验药械相关的资料。
六、药械临床试验部门保证所拥有的软硬件设施满足安全有效地进行临床试验的需要,确保所有研究者都具备承担该项临床试验的专业特长、资格和能力,并经过培训。临床试验开始前,研究者和申办者应就试验方案、试验监查、标准操作规程以及试验中的职责分工等达成书面协议。书面协议一式三份,申办方,研究者和科教科各执一份。
七、 在临床试验中研究者须对受试者在医疗上认真负责: 在临床试验前对每一名受试者的医学情况进行全面检查,包括其诊断、合并症、用药情况;在临床试验中密切观察任何与试验相关的不良事件,包括异常的实验室检查值等。如发生不良事件应给予及时处理;试验结束后继续观察一段时间,注意可能延迟出现的不良反应并予以处理。
1
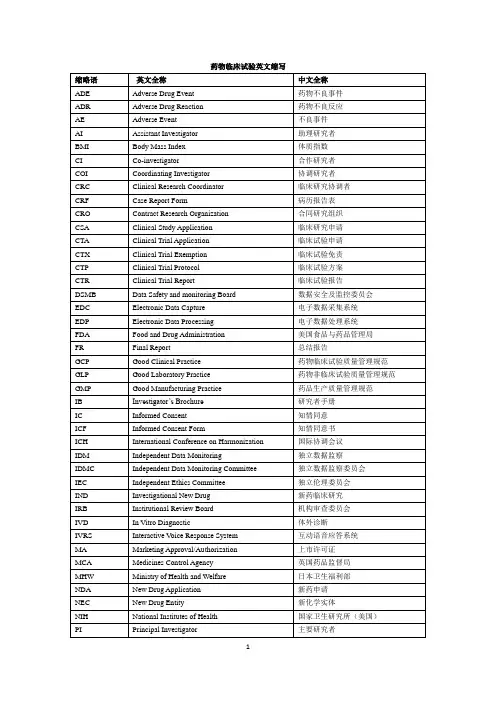
药物临床试验英文缩写
缩略语 英文全称 中文全称
ADE Adverse Drug Event 药物不良事件
ADR Adverse Drug Reaction 药物不良反应
AE Adverse Event 不良事件
AI Assistant Investigator 助理研究者
BMI Body Mass Index 体质指数
CI Co-investigator 合作研究者
COI Coordinating Investigator 协调研究者
CRC Clinical Research Coordinator 临床研究协调者
CRF Case Report Form 病历报告表
CRO Contract Research Organization 合同研究组织
CSA Clinical Study Application 临床研究申请
CTA Clinical Trial Application 临床试验申请
CTX Clinical Trial Exemption 临床试验免责

药物临床试验结束临床试验的标准操作规程
药物临床试验结束阶段仍需按照GCP的要求,规范各个环节的操作,为此应制定结束临床试验标准操作规程,其主要内容包括:
(一)试验用药物的处理
1.专业科室试验药物管理员将“试验用药物登记表”等交机构办公室归档。
2.机构办公室药物管理人员清点所有剩余试验用药及应急信封,返还申办者或与申办者共同销毁,并填写“临床试验剩余药物销毁/退回记录表”,双方共同签名,一式两份,其中一份交机构办公室归档。
(二)分中心小结
临床试验的主要研究者根据本研究中心的数据资料按要求格式撰写分中心小结,并签名及日期。
(三)揭盲规程
1.揭盲时间 全部临床试验完成,资料收集齐全并全部上交到组长单位后。
2.执行揭盲人员 申办者、组长单位主要研究者、统计负责人员等。
3.揭盲地点 组长单位指定的地点。
4.揭盲程序 分两级揭盲。数据库锁定、统计资料前进行一级揭盲,只揭出受试者接受的不同治疗组别,即A、B组。统计结束、得到分析结果后进行二级揭盲,揭出A、B组的具体分组情况,即治疗组和对照组。
(四)统计
1.进一步审核CRF表:数据管理员审核经监查员、研究者签名的CRF表,必要时填写质询表。
2.按临床试验数据统计处理SOP进行数据统计处理。
(五)总结
临床试验组长单位的主要研究者应写出试验总结报告,经核实确认后各中心主要研究者应签署“研究者签名表”。
(六)资料归档
临床试验研究者填写“完成试验受试者编码目录”表格,整理全部临床试验资料、归档。机构办公室根据《药物临床试验质量管理规范》的规定审查归档资料和病例报告表,确认病例报告表已经过临床试验负责人签字,原始住院病历归档后,在病例报告表上盖章,并将“临床试验保存文件”逐一登记。
(七)审核盖章
机构办公室根据有关规定,审核临床试验总结或小结,盖章后交给申办者。
一、制定伦理委员会标准操作规程的标准操作规程建立制定伦理委员会标准操作规程的标准操作规程目的是为了保证各项标准操作规程的制定科学合理、更新及时和操作规范,适用于伦理委员会的各项工作。 (一)制定标准操作规程应涵盖的范围

Accuracy 准确度
Active control, AC 阳性对照,活性对照
Adverse drug reaction, ADR 药物不良反应
Adverse event, AE 不良事件
Adverse medical events 不良医学事件
Adverse reaction 药物不良反应
Alb 白蛋白
ALD(Approximate Lethal Dose) 近似致死剂量
ALP 碱性磷酸酶
Alpha spending function 消耗函数
ALT 丙氨酸氨基转换酶
Analysis sets 统计分析的数据集
Approval 批准
Assistant investigator 助理研究者
AST 天门冬酸氨基转换酶
ATR 衰减全反射法
AUCss 稳态血药浓度-时间曲线下面积
Audit 稽查
Audit or inspection 稽查/视察
Audit report 稽查报告
Auditor 稽查员
Bias 偏性,偏倚
Bioequivalence 生物等效应
Blank control 空白对照 Blind codes 编制盲底
Blind review 盲态审核
Blind review 盲态检查
Blinding method 盲法
Blinding/ masking 盲法,设盲
Block 分段
Block 层
Block size 每段的长度
BUN 尿素氮
Carryover effect 延滞效应
Case history 病历
Case report form 病例报告表
Case report form/ case record form, CRF 病例报告表,病例记录表
Categorical variable 分类变量
Cav 平均浓度
CD 圆二色谱
CL 清除率
Clinical equivalence 临床等效应
Clinical study 临床研究