氰乙基对几种芳胺结构和光谱的影响_唐智勇
- 格式:pdf
- 大小:433.26 KB
- 文档页数:6



第43卷第4期质谱学报Vol.43 No.4 2022年7月JournalofChineseMassSpectrometrySocietyJul.2022合成大麻素类毒品的结构衍变与质谱解析魏紫薇1,王潇逸1,李 珊1,覃仕扬2,辛国斌2,王元凤1,2(1.中国政法大学证据科学研究院,北京 100088;2.法庭毒物学公安部重点实验室,北京 100192)摘要:合成大麻素毒品蔓延趋势严峻,我国政府启动了整类列管政策。
该类毒品结构变异较强,对重点目标结构的识别以及共性质谱特征的汇总在司法实践中具有重要意义。
本文解析了该类毒品的结构衍变规律,遴选27种合成大麻素,采用气相色谱 质谱联用法(GC MS)采集EI MS数据。
实验结果表明,合成大麻素类物质主要由母核、链接、取代基和侧链四部分构成。
其中,吲哚/吲唑母核易产生犿/狕144、145特征碎片;链接部分的碎裂主要发生在羰基两侧;取代基上的叔碳原子、丁酸甲酯以及丁酰胺易发生多元化碎裂;侧链易发生中性丢失而产生氟丁基(犿/狕74)、氟戊基(犿/狕88)和戊烯基(犿/狕68)等特征碎片。
该碎裂途径的推断可为未知新型合成大麻素类物质的判别提供参考。
关键词:甲酰基吲哚类;甲酰胺基吲哚/吲唑类;合成大麻素;气相色谱 质谱(GC MS)中图分类号:O657.63 文献标志码:A 文章编号:1004 2997(2022)04 0482 13犱狅犻:10.7538/zpxb.2021.0173犛狋狉狌犮狋狌狉犪犾犇犲狏犲犾狅狆犿犲狀狋犪狀犱犕犪狊狊犛狆犲犮狋狉狅犿犲狋狉狔犃狀犪犾狔狊犻狊狅犳犛狔狀狋犺犲狋犻犮犆犪狀狀犪犫犻狀狅犻犱狊WEIZi wei1,WANGXiao yi1,LIShan1,QINShi yang2,XINGuo bin2,WANGYuan feng1,2(1.犐狀狊狋犻狋狌狋犲狅犳犈狏犻犱犲狀犮犲犔犪狑犪狀犱犉狅狉犲狀狊犻犮犛犮犻犲狀犮犲,犆犺犻狀犪犝狀犻狏犲狉狊犻狋狔狅犳犘狅犾犻狋犻犮犪犾犛犮犻犲狀犮犲犪狀犱犔犪狑,犅犲犻犼犻狀犵100088,犆犺犻狀犪;2.犓犲狔犔犪犫狅狉犪狋狅狉狔狅犳犉狅狉犲狀狊犻犮犜狅狓犻犮狅犾狅犵狔,犕犻狀犻狊狋狉狔狅犳犘狌犫犾犻犮犛犲犮狌狉犻狋狔,犅犲犻犼犻狀犵100192,犆犺犻狀犪)犃犫狊狋狉犪犮狋: Theabuseofnewpsychoactivesubstanceshasspreadaroundtheworld.Syn theticcannabinoids(SCs)isanimportantgroup.SincethefirstdiscoveryofJWH 018inEuropein2008,SCshavemultipliedrapidlyandbecomethefocusofdrugcontrolworldwide.StatisticsfromtheDrugIntelligenceandForensicCenteroftheMinistryofPublicSecurityofChinashowedthattheproportionofSCsinsuspecteddrugsamplesseizedhadincreasedfrom18.5%to72%in2017 2019.Tothis,ourcountrydrugcon troladministrationdepartmentmakespositiveresponse.Aseriesofnewanti drugpoli cieshadbeenintroducedsince2015.Fifty threeSCswerelegallycontrolleduntiltheendof2018.ChinahadissuedtotalbanonSCssinceJuly1st,2021.However,becauseofthegreatstructuralvariabilityofSCs,itisofgreatsignificancetoidentifythekeytargetstructuresandsummarizethecharacteristicsofthecommonspectrumofsimilardrugsin国家社会科学基金项目(19BFX195);国家重点研发计划项目(2018YFC0807304)本文通信作者王元凤judicialpractice.Inthispaper,27syntheticcannabinoidswereselected,includingthenewsyntheticcannabinoidswithhighprevalenceinrecentyearsandpotentialdrugstructuresthathadnotbeenreportedbutwereinferredbasedonthestructuralevolutiontrendofsyntheticcannabinoids.Themethodofgaschromatography massspectrometry(GC MS)wasusedtocollectdataoftheselectedSCsunderelectronionizationmode(EI).TheresultsindicatedthattheSCsweremainlycomposedofcore,linker,linkedgroupandsidechain.Amongthem,thecoreofnewSCsgraduallychangedfromformylindoletoindole/indazolamide,whichwouldproducecharacteristicfragmentsof犿/狕144,145,respectively.Thefragmentationofthelinkerpartmainlyoccurredonbothsidesofthecarbonylgroup.Thetertiarycarbonatoms,methylbutyrateandbutylamidefromthelinkedgroupsledtodiversifiedfragmentationeasily.Neutrallossfrequentlyhappenedinthepartofsidechain,whichresultedintheformationofcharacteristicfrag mentssuchasfluoro butyl(犿/狕74),fluoro pentyl(犿/狕88)andpentenyl(犿/狕68).The7SCspreparedaccordingtothepredictedstructuresharedsimilarfragmentationpathwayswiththe22SCsabusedbothdomesticallyandabroad.ThevariationofSCsbasedonthestructuresdefinedinlawwouldgraduallydecreaseandthestructuralstabil itywouldbeenhancedafterthetotalbanofSCswasissued.ThemassfragmentationpathwaymentionedabovecanprovidetechnicalsupportforthespeciesidentificationofnewSCs,aswellasthepreventionofdrugvariation.犓犲狔狑狅狉犱狊:formylindole;indole/indazolecarboxamide;syntheticcannabinoids;gaschromatography massspectrometry(GC MS) 新精神活性物质的滥用已席卷全球[1 3]。

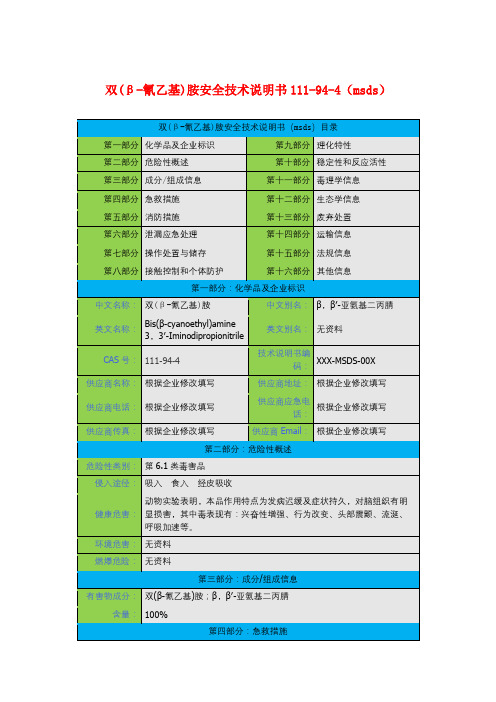

氰乙基结构式摘要:一、氰乙基结构式简介1.氰乙基的定义2.氰乙基的结构式二、氰乙基的性质与应用1.氰乙基的物理性质2.氰乙基的化学性质3.氰乙基的应用领域三、氰乙基的安全措施与注意事项1.氰乙基的毒性2.氰乙基的防护措施3.氰乙基的储存与运输四、氰乙基的发展趋势与前景1.氰乙基的研究进展2.氰乙基的新应用领域3.氰乙基的未来发展前景正文:氰乙基是一种有机化合物,其化学式为C2H2N3,结构式中含有一个氰基(-CN)和一个乙基(-CH2CH3)基团。
氰乙基广泛存在于许多化合物中,具有一定的物理性质和化学性质,并在许多领域中有着广泛的应用。
一、氰乙基结构式简介氰乙基是由一个氰基(-CN)和一个乙基(-CH2CH3)基团组成的有机化合物。
其结构式为:CH2CNCH3。
氰乙基是一种重要的有机化工原料,广泛应用于农药、医药、染料等领域。
二、氰乙基的性质与应用氰乙基具有以下物理性质:无色至微黄色液体,有刺激性气味;熔点-14.6℃,沸点105.6℃;闪点42.2℃;相对密度0.914g/cm。
氰乙基的化学性质活泼,容易与其他物质发生反应,特别是在碱性条件下,容易发生亲核加成反应。
氰乙基可以与醇、胺、卤代烷等化合物发生反应,生成各种氰乙基衍生物。
氰乙基的应用领域非常广泛,主要包括农药、医药、染料、橡胶助剂等。
在农药领域,氰乙基作为合成中间体,可生产多种杀虫剂、杀菌剂等;在医药领域,氰乙基可用于合成多种药物,如抗病毒药物、抗肿瘤药物等;在染料领域,氰乙基可用于合成各种染料和颜料;在橡胶助剂领域,氰乙基可作为硫化促进剂等。
三、氰乙基的安全措施与注意事项氰乙基具有一定的毒性,对人体和环境有潜在的危害。
氰乙基的急性中毒症状包括头痛、恶心、呕吐、腹泻、抽搐、昏迷等,严重时可能导致死亡。
长期接触氰乙基,可能导致慢性中毒,影响神经系统、肝脏、肾脏等器官的功能。
为了保证人身安全和环境保护,在处理氰乙基及其衍生物时,应采取以下安全措施:1.佩戴防护装备,如口罩、手套、护目镜等;2.保持操作场所通风良好;3.避免与酸、碱等强氧化剂接触;4.氰乙基泄漏时,应立即采取措施进行处理;5.氰乙基的储存、运输应符合相关法规要求。
氰乙基对几种芳胺结构和光谱的影响唐智勇1胡云楚1,*赵莹1刘述斌2,*(1中南林业科技大学应用化学研究所,长沙410004;2Research Computing Center,University of North Carolina,Chapel Hill,North Carolina 27599-3420,USA)摘要:采用量子化学中密度泛函理论(DFT)的B3LYP 方法分别用6-31G *和6-311+G *基组对苯胺、对氯苯胺和对甲苯胺及其氰乙基衍生物的几何构型进行全优化,探讨了氨基上氰乙基的引入对分子电荷转移、前线轨道能量和电子光谱等性质的影响规律.在此基础上采用含时密度泛函方法(TD -DFT)计算了分子第一激发态的电子跃迁能,得到最大吸收波长λmax .计算结果表明,氨基上氰乙基的引入,对前线分子轨道组成影响虽然小,但使得最大吸收波长红移,与实验值λmax 有较好的一致性,发现该类物质主要吸收光谱源于分子内的π→π*的电子跃迁.关键词:芳胺;氰乙基;密度泛函理论;前线轨道;电子光谱中图分类号:O641Impact of Cyanoethy Groups on Structure and Spectroscopy of a FewAromatic AminesTANG Zhi -Yong 1HU Yun -Chu 1,*ZHAO Ying 1LIU Shu -Bin 2,*(1Institute of Applied Chemistry,Central South University of Forestry &Technology,Changsha410004,P.R.China ;2Research Computing Center,University of North Carolina,Chapel Hill,North Carolina27599-3420,USA )Abstract :The impact of introducing cyanoethy groups in aromatic amines on structure,charge transfer,frontier orbital energy and composition,as well as electronic absorption spectra was investigated in this work.Theoretical studies of aniline,p -chloroaniline,p -toluidine,and cyanoethy derivatives were carried out at density functional theory (DFT)B3LYP/6-31G *and B3LYP/6-311+G *levels to obtain optimized geometrical structures.Time -dependent density functional theory was applied to calculate the first excited state electronic transition energy and maximum absorption wavelength λmax .Our results indicate that the introduction of a cyanoethy group has limited effect on the composition of frontier orbitals.The main absorption spectrum originates from the featured π→π*electronic transition.The predicted spectra agree well with the available experimental findings.Key Words :Aromatic amine;Cyanoethy;Density functional theory;Frontier orbital;Electronic spectrum[Article]物理化学学报(Wuli Huaxue Xuebao )Acta Phys.-Chim.Sin .,2009,25(4):701-706Received:October 29,2008;Revised:December 15,2008;Published on Web:February 16,2009.*Corresponding authors.Email:hucsfu@,shubin@;Tel:001-919-962-4032.2009年度湖南省研究生创新基金和2008年度中南林业科技大学研究生创新基金资助项目鬁Editorial office of Acta Physico -Chimica Sinica芳香胺的氰乙基化反应是有机合成中最基本的反应之一,其氰乙基化产物是分散染料、医药、表面活性剂的重要中间体,可广泛用于染料、抗氧化剂、抗菌剂、表面活性剂等精细化学品的合成.Allen 等[1]采用CuOAc ·H 2O 作催化剂,系统研究了10多种芳胺的单氰乙基化反应,其单氰乙基产物的收率大多小于80%,其中对氯苯胺的单氰乙基收率为78%;钟为慧等[2]采用SmI 3作催化剂研究了5种活性高的芳胺氰乙基化反应,单氰乙基化收率为59%-83%;郑立红等[3]采用CuCl 作催化剂,利用邻氯苯胺循环来合成N -氰乙基邻氯苯胺,其收率接近75%.总之,从国内外对芳胺的单氰乙基化反应的April 701Acta Phys.-Chim.Sin.,2009Vol.25研究结果来看,未有一种催化剂能使反应的转化率达到90%以上.而对于芳胺的双氰乙基化反应,有关报道却很少.仅有Braunholtz 等[4]以HAc -CuCl 催化体系反应16h 合成N ,N -二氰乙基对氯苯胺,收率只有20%.可见,要获得双氰乙基物较难.因此工业上双氰乙基产物的获得一般采用分离单、双氰乙基产物的办法.近几年来,赵莹等[5-9]开展了一系列用无水AlCl 3催化芳胺与丙烯腈进行加成反应的研究工作,合成出10多种氰乙基加成产物,收率达76%-98%,有的已经实现工业化生产并被用来合成分散染料.然而对于芳香胺的氰乙基化产物的理论研究尚未见报道.目前,应用于激发态能量计算的理论方法有多种[10],而用含时密度泛函方法(TD -DFT)[11-19]处理电子激发态问题成为当前一个非常活跃的研究领域.本研究运用Guassian 03量子化学程序包[20]中的密度泛函理论(DFT)[21-23],采用B3LYP [24,25]方法分别在6-31G *和6-311+G *水平下对合成的一系列芳香胺的氰乙基化产物进行了量子化学计算,从理论上预测氨基上氰乙基的引入对芳胺分子电荷转移、前线轨道能量和电子光谱等性质的影响规律,探讨此类分子中第一激发态电子跃迁的本质,并通过实验验证了几种芳胺及氰乙基芳胺光谱规律,从而为设计新型染料结构提供理论依据.1计算模型和方法运用Guassian 03量子化学程序包[20]中的密度泛函理论[21-23],首先在B3LYP/6-31G *和B3LYP/6-311+G *水平上,对几种芳香胺及其氰乙基衍生物(图1)进行全自由度几何优化,并做频率分析,频率分析结果均无虚频,表明选用的目标化合物的优化结果确实为其稳定构型.以基态的优化构型为基础,应用含时密度泛函理论TD -DFT [11-19],分别在相同理论水平(B3LYP/6-31G *和B3LYP/6-311+G *)上计算化合物的电子吸收光谱,并与实验结果进行比较,计算中所采用的收敛精度为程序设定值.将6-31G *和6-311+G *两个基组得到的结果进行比较,发现几何构型、净电荷布居和前线轨道数据相差甚微,不影响全文对结构的分析,而在最大吸收波长方面有较大的差别,所以,结果与讨论中的前3节只列出了6-31G *水平下的数据,在第4节中讨论了两种水平下的最大吸收波长.2结果与讨论2.1几何构型表1给出了在6-31G *水平下9种芳胺分子的部分优化键长、键角和二面角.从表1可以看出,L (6,7)的C —Cl 键长均小于饱和卤代烷烃中C —Cl 键长(0.177nm)[26],原因是由于氯原子的p 轨道与苯环上π键有部分重叠,因此,C —Cl 键之间也有部分双键的性质,使得C —Cl 键长变短;L (6,7)的C —C 键长均介于C —C 单键(0.154nm)[26]和C 襒C 双键(0.134nm)[26]之间,表明具有一定的双键性质,引入氰乙基后,对L (6,7)的C —Cl 和C —C 几乎没有影响,原因是氰乙基处于苯胺分子的对位,距离较远;777991010R1=H,CH 3,Cl;R2=H,CH 2CH 2CN;R3=H,CH 2CH 2CN图1芳香胺及其衍生物的分子结构模型和原子编号Fig.1Structure model and atomic number ofaromatic amine and its derivativesL :bond length in nm;D :dihedral angle in degree;X:CH 2CH 2CN;optimized at B3LYP/6-31G *level表1芳香胺及其衍生物的几何构型及其优化参数Table 1Optimized geometries and structural characteristics of aromatic amine and its derivativesCompoundA1A2A3B1B2B3E1E2E3R1H H H Cl Cl Cl CH 3CH 3CH 3R2H H X H H X H H X R3H X X H X X H X X L (6,7)0.10870.10860.10860.17610.17610.17580.15110.15110.1511L (3,8)0.14460.14020.14050.14430.14000.14020.14020.14010.1406L (8,9)0.10200.10150.14500.10190.10140.14560.10130.10130.1450L (8,10)0.10200.14500.14500.10190.14520.14510.10130.14480.1450D (4,5,6,7)-180.00179.72-179.68180.00179.69179.71-178.59179.68178.45D (1,2,3,8)180.00-177.91178.72180.00-177.88-179.06-177.22178.20178.38D (2,3,8,9)-58.04-27.043.43-58.28-26.816.11-155.9425.042.16D (2,3,8,10)58.08-164.48179.8258.41-165.80177.95-26.80165.59178.57702No.4唐智勇等:氰乙基对几种芳胺结构和光谱的影响calculated at B3LYP/6-31G *levelL (3,8),L (8,9)和L (8,10)的C —N 键长均介于C —N 单键(0.147nm)[26]和C 襒N 双键(0.127nm)[26]之间,表明也具有一定的双键性质,与苯环形成共轭.几何构型全优化后,系列苯胺分子的主要骨架其二面角均接近于-180°或180°,氨基上两个氢原子都被氰乙基取代后,与氨基相连氰乙基碳的二面角接近于0°和180°,说明氰乙基引入后,导致氨基构型发生扭转,分子平面延伸.苯胺、对氯苯胺和对甲苯胺分子结构中主要原子和原子团基本能保持在同一平面上.故这些原子和苯环一起构成了大的共轭体系.2.2净电荷布居表2给出了在6-31G *水平下9种芳胺分子部分原子的净电荷布居,从表2可以看出:氰乙基取代后的几种苯胺分子发生了不同程度的电荷转移,与其直接键连的原子的电荷分布变化较大,离取代基越远,原子上的电荷分布变化越小.化合物B1由于Cl 的吸电子诱导效应大于p -π共轭效应,使得C(6)上净电荷明显降低;而化合物E1苯环上所有C 的净电荷都明显升高,尤其是C(6),所带的电荷由负变正,说明甲基主要以p -π共轭效应为主,电子云向相邻碳转移.氨基与苯环形成p -π共轭,由于氰乙基的吸电子诱导效应,引入后使得氨基N 原子上负的净电荷明显降低,程度顺序为双氰乙基>单氰乙基;C(3)上正的净电荷明显增加,程度顺序为双氰乙基>单氰乙基,对位上有吸电基Cl 或供电基CH 3几乎无影响.化合物的反应活性与分子中原子所带净电荷的多少有关,电荷集中的原子就是分子反应的活性中心.原子的正电荷越多,受亲核试剂进攻的可能性越大;反之,原子的负电荷越多,其受亲电试剂进攻的可能性越大.因此,从表2中可以看出,负电荷主要集中在氨基N(8)上,可以预测N(8)是亲电反应的作用点,有利于亲电试剂丙烯腈进攻.正是由于N 原子上负电荷远大于苯环上各碳原子,才使得几种苯胺与丙烯腈反应,生成的是N -氰乙基苯胺和N ,N -二氰乙基苯胺,而不是苯环上氰乙基取代苯胺,这点在文献[1-5]中已得到证实.2.3前线轨道能量量子化学的前线轨道理论认为,分子参与反应则分子轨道要发生变化,优先起作用的是前线轨道,即分子中的最高占有轨道(HOMO)和最低空轨道(LUMO).对分子轨道的分析可以为我们提供很多有关分子的信息,比如电子的结合位置及分子的化学反应性能[27-29],此外,系列苯胺分子的前线轨道能量与光电子能谱密切相关,而前线轨道能量差与紫外-可见光谱性质密切相关.表3给出了在6-31G *水平下9种芳胺分子的表3芳香胺及其衍生物前线轨道能级(a.u.)Table 3Frontier molecular orbital energy levels (a.u.)of aromatic amine and its derivatives表2芳香胺及其衍生物部分原子的净电荷布居(e )Table 2Net charge populations (e )of aromatic amine and its derivativesCompoundA1A2A3B1B2B3E1E2E3C(1)-0.138-0.134-0.137-0.139-0.135-0.140-0.188-0.184-0.192C(2)-0.156-0.188-0.197-0.152-0.183-0.189-0.170-0.184-0.195C(3)0.2060.3500.3570.2100.3570.3620.3020.3490.354C(4)-0.125-0.183-0.197-0.121-0.177-0.190-0.170-0.179-0.195C(5)-0.140-0.138-0.137-0.140-0.141-0.140-0.188-0.196-0.190C(6)-0.126-0.136-0.133-0.061-0.071-0.0680.1750.1790.182C(7)-0.529-0.531-0.531Cl(7)-0.029-0.034-0.026N(8)-0.778-0.668-0.489-0.779-0.670-0.490-0.787-0.654-0.491C(9)-0.157-0.158-0.156C(10)-0.112-0.157-0.111-0.161-0.102-0.156Compound A1A2A3B1B2B3E1E2E3HOMO -0.2404-0.2034-0.2147-0.2414-0.2126-0.2171-0.1922-0.1973-0.2086LUMO 0.00130.0025-0.0148-0.0139-0.0159-0.02630.00980.0035-0.0137ΔE0.24170.20590.19990.22750.19670.19080.20200.20070.1949calculated at B3LYP/6-31G *level703Acta Phys.-Chim.Sin.,2009Vol.25前线轨道能级.从表3可以看出,对位Cl 取代使分子的前线轨道能降低,对位CH 3取代使分子的前线轨道能量升高.取代基的引入使分子能隙变小,供电子基CH 3对能隙的减小更为明显,氰乙基的引入及数目增加对减小能隙起促进作用.能隙大小顺序为苯胺>单氰乙基苯胺>双氰乙基苯胺.ΔE 随氰乙基的引入逐渐变小,表明吸收光谱的最大吸收峰可能会出现红移现象.计算得到的能隙大小顺序,预示着系列苯胺分子电子光谱最大吸收波长的大小顺序.从HOMO 轨道和LUMO 轨道的能量上看,能量变化较大,它们是反应的主要参与轨道.为了较全面地利用轨道理论来解释分子反应活性,画出了几种芳香胺及其氰乙基衍生物的最高占有轨道(HOMO)和最低空轨道(LUMO)的空间分布图(图2).分别将化合物分子中的所有原子的轨道组合为分子轨道系数的平方和,归一化后,得到各原子在前线分子轨道及附近分子轨道中的贡献(表4).图2给出了在6-31G *水平下9种芳胺分子的前沿分子轨道图形,从中可以看出,9种芳香胺的HOMO 和LUMO 轨道都是π分子轨道.表4给出了在6-31G *水平下9种芳胺分子HOMO 和LUMO 中主要原子的贡献百分率(为便于比较,小于1.5的数值忽略不计),从表4可以看出,化合物B1的表4前线轨道中各主要原子的贡献百分率(%)Table 4Main atomic contributive percents (%)of frontier orbital for molecular orbital图2芳香胺及其衍生物的前沿分子轨道Fig.2Frontier molecular orbitals of aromatic amine and its derivativesMain atomC(1)C(2)C(3)C(4)C(5)C(6)Cl/C(7)N(8)C(9)C(10)A1HOMO 17.82 6.6728.0310.17 4.9327.75 1.78LUMO 5.6032.6812.60 4.5731.8011.34-A2HOMO 5.6210.899.589.71 4.4617.8637.59 3.07LUMO 20.7727.72-19.7527.50---A3HOMO 6.9110.437.0410.43 2.4316.7137.29--LUMO 20.4118.46-18.8920.42-- 3.303.40B1HOMO 14.46 4.6819.53 4.99 6.1918.9526.70 1.64LUMO 6.9930.778.69 6.6129.968.76--B2HOMO 6.299.059.218.44 3.4915.9112.1031.35-LUMO 21.6227.81-19.8927.61----B3HOMO 8.119.11 6.858.57 2.3915.1411.1331.51--LUMO 23.7222.60-21.8723.72-----E1HOMO 7.2110.4312.3610.44 2.0619.77-31.10LUMO 22.3121.81-21.8221.80---E2HOMO 7.469.339.9410.28 2.1216.85-34.39-LUMO 25.9225.35-22.5223.94----E3HOMO 8.2010.449.629.85 1.8317.11-37.39--LUMO22.7322.19-20.2625.35-----calculated at B3LYP/6-31G *level704No.4唐智勇等:氰乙基对几种芳胺结构和光谱的影响λmax in nm;a)excitation energies at the TD-DFT/6-31G*level,b)oscillator strengths at the TD-DFT/6-31G*level,c)excited state,d)calculated at the TD-DFT/6-31G*level;e)calculated at the TD-DFT/6-311+G*level,f)experimental data were determined in the solvent of ethanol.HOMO电子云主要集中在苯环和对位Cl(7)上,其中Cl(7)占26.70%,与之相连的C(6)占18.95%,而N(8)上电子分布几乎为0,其原因是,虽然Cl(7)和N(8)都与苯环形成了p-π共轭,但是Cl的电负性大于N,吸电子诱导效应大于共轭效应,所以电子云主要集中于C(6)和Cl(7)上;化合物E1的HOMO电子云主要集中在苯环和N(8)上,其中N(8)占31.10%,而对位CH3上接近于0,这是因为CH3是推电子基,使得电子云主要离域在N(8)周围;化合物A2、A3、B2、B3、E2和E3的HOMO电子云主要分布在N(8)和苯环上,其中N(8)都占31%以上,主要是N 的电负性大于C和氰乙基的吸电子诱导效应.9个化合物的LUMO轨道电子云都分散在苯环上.从图2中能更加清晰地看出其前线轨道组成特征.由此可以预测对氯苯胺难与丙烯腈发生亲电取代反应,而对甲苯胺易与丙烯腈发生亲电取代反应.通过实验也证实了这点,在无水AlCl3催化下,前者回流反应24h,产率只有85%左右[6];后者回流反应12h,产率有93%以上[5].2.4电子吸收光谱近年来,TD-DFT方法已越来越多地被应用于有机化合物光谱性质的研究.我们采用UV-1100紫外-可见分光光度计以无水乙醇为溶剂,分别测定了苯胺、对氯苯胺和对甲苯胺及其氰乙基衍生物9种化合物的紫外-可见光谱,它们的最大吸收波长列于表5中.表5给出了几种芳胺及其氰乙基衍生物在6-31G*和6-311+G*水平下的最大吸收波长.从表5中可以看到,系列苯胺分子的最大吸收波长计算值和实验值之间有一定差异,可能在于程序计算是按理想气态分子处理的,而实验值是以无水乙醇为溶剂获得的,但仍有着较好的一致性.除化合物A1和B1外, 6-31G*水平下λmax大都比实验λmax小约38nm,而6-311+G*水平下λmax基本上比实验λmax小约24nm,由此可知,6-311+G*基组在分析小分子芳胺光谱性质上比6-31G*基组相对准确些,从系列苯胺分子的最大吸收波长的跃迁能量顺序来看,与表3的能隙顺序一致.系列苯胺分子的最大吸收波长均位于近紫外区,λmax计算值按—NH2、—NHCH2CH2CN、—N(CH2CH2CN)2顺序红移,这与实验值一致.计算结果较好地反映实验的变化规律.3结论对自行合成的系列苯胺分子进行了量子化学计算,从理论上预测了氨基上—CH2CH2CN的引入对分子电荷转移、前线分子轨道能量和电子吸收光谱等性质的影响规律.通过理论计算并与实验结果比较发现,采用DFT-B3LYP方法研究简单苯胺分子及其氰乙基衍生物可以得到较合理的几何构型和电子吸收光谱结果.通过对分子中主要原子净电荷的分析,揭示了诱导效应和共轭效应影响9种苯胺分子电荷分布,由主要原子所带电荷多少知道几种苯胺分子的反应活性中心以及与亲电试剂丙烯腈反应的难易程度.通过对前线轨道的分析,认为9种苯胺分子的HOMO和LUMO均为π分子轨道,其主要由氨基和苯环所贡献.分子轨道研究表明,此类化合物最大吸收波长是分子吸收能量之后电子从HOMO向LUMO的跃迁,即电子从共轭体系中给电子体跃迁到受电子体引起的.取代基的引入,均导致最大吸收波长红移,吸电子基引起的红移程度大于供电子基,理论计算与实验结果的规律基本一致.根据化学结构和电子吸收光谱可以预测这一系列苯胺分子的紫外吸收区,从而为染料的合成,如测定其敏化区的位置提供帮助.了解芳胺类分子电子结构和光谱性质是理解其作为光敏材料、染料中间体和进行分子设计的起点.故在DFT框架内表5芳香胺及其衍生物的λmax计算值与实验值的比较Table5Comparison of calculated and experimental data ofλmax for aromatic amine and its derivatives Compound A1A2A3B1B2B3E1E2E3E a 5.4704 5.3312 4.7244 5.2713 4.6728 4.5231 4.8079 4.7698 4.6257f b0.00210.02600.03660.00730.03200.03360.03280.03000.0376 Characterπ→π*π→π*π→π*π→π*π→π*π→π*π→π*π→π*π→π* Transition c25→2639→4053→5429→3043→4457→5833→3447→4861→62λmax(calc.)d226.64252.56262.44235.21265.33274.11257.88259.94268.03λmax(calc.)e232.42267.09271.07242.11278.18284.26271.34273.96278.52λmax(expt.)f278.5288.5295.5300.0306.0310.0293.0298.0301.0705Acta Phys.-Chim.Sin.,2009Vol.25进行结构与性质的研究,尤其是应用含时密度泛函理论对此类芳胺中间体作单电子跃迁的计算,将为以后改构设计分子提供参考依据.References1Allen,.Chem.,1957,22:12132Zhong,W.H.;Zhang,.Chem.,2000,20(5): 747[钟为慧,张永敏.有机化学,2000,20(5):747]3Zheng,L.H.;Wang,X.H.;Zhao,C.Y.Dyestuff Industry,2001,4: 26[郑立红,王宪花,赵从伊.染料工业,2001,4:26]4Braunholtz,J.T.;Mann,F.G.J.Chem.Soc.,1953:18175Zhao,Y.;Tan,X.Y.;Yang,Z.;Li,.Chem.,2005, 25(11):1469[赵莹,谭晓艳,杨志,李姣娟.有机化学,2005,25(11):1469]6Zhao,Y.;Ye,C.C.;Li,J.J.;Yang,Z.Chemistry,2005,69(6):438 [赵莹,叶翠层,李姣娟,杨志.化学通报,2005,69(6):438] 7Gong,J.L.;Li,J.J.;Zhao,Y.Dyestuffs and Coloration,2008,45(1):44[龚建良,李姣娟,赵莹.染料与染色,2008,45(1):44] 8Zhao,Y.;Ye,C.C.Chemical Reagents,2006,28(5):81[赵莹,叶翠层.化学试剂,2006,28(5):81]9Zhao,Y.;Tan,X.Y.;Li,J.J.;Yang,Z.Chin.J.Syn.Chem.,2006, 14(3):297[赵莹,谭晓燕,李姣娟,杨志.合成化学,2006, 14(3):297]10Liu,S.F.;Seward,C.;Aziz,H.;Hu,N.X.;Popovic,Z.;Wang,S.anometal.,2000,19(26):570911Belletete,M.;Durocher,G.;Hamel,S.;Cote,M.;Wakim,S.;Leclerc,M.J.Chem.Phys.,2005,122(10):4303112Fan,J.X.;Ren,A.M.;Feng,J.K.;Bo,D.S.Chem.J.Chin.Univ., 2006,27(6):1091[范建训,任爱民,封继康,薄东生.高等学校化学学报,2006,27(6):1091]13Xu,H.;Yang,B.;He,F.;Xie,Z.Q.;Tian,L.L.;Liu,X.D.;Yu,J.S.;Ma,Y.G.Chem.J.Chin.Univ.,2006,27(3):510[许海,杨兵,何凤,解增旗,田雷蕾,刘晓冬,于景生,马於光.高等学校化学学报,2006,27(3):510]14Li,H.X.;Xiao,T.Chem.J.Chin.Univ.,2007,28(4):747 [李会学,萧泰.高等学校化学学报,2007,28(4):747]15Liu,J.N.;Chen,Z.R.;Yuan,S.F.Acta Phys.-Chim.Sin.,2005, 21(4):402[刘军娜,陈志荣,袁慎峰.物理化学学报,2005,21(4):402]16Liang,X.Q.;Pu,X.M.;Shu,Y.J.;Tian,A.M.Acta.Chim.Sin., 2006,64(20):2057[梁晓琴,蒲雪梅,舒远杰,田安民.化学学报,2006,64(20):2057]17Chen,Q.W.;Wang,L.Y.;Zhai,G.H.;Wen,Z.Y.;Zhang,Z.X.Acta Chim.Sin.,2005,63(1):39[陈沁闻,王兰英,翟高红,文振翼,张祖训.化学学报,2005,63(1):39]18Xue,Y.S.;Gong,X.D.;Xiao,H.M.;Tian,H.Acta Chim.Sin., 2004,62(10):963[薛运生,贡雪东,肖鹤鸣,田禾.化学学报, 2004,62(10):963]19Runge,E.;Gross,E.K.U.Phys.Rev.Lett.,1984,52:99720Frish,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian03, Revision B.02.Pittsburgh PA:Gaussian,Inc.,200321Marques,M.A.L.;Gross,E.K.U.Annu.Rev.Phys.Chem.,2004, 55:42722Dreuw,A.;Head-Gordon,M.Chem.Rev.,2006,105:4009.23Huang,Y.;Zhong,A.G.;Rong,C.Y.;Xiao,X.M.;Liu,S.B.J.Phys.Chem.A,2008,112:30524Becke,A.D.J.Chem.Phys.,1992,97:917325Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B,1988,37:78526Tao,S.X.;Zhao,J.W.Advanced organic chemistry.Beijing: People′s Education Press,1981:16[陶慎熹,赵景文.高等有机化学.北京:人民教育出版社,1981:16]27Liu,S.B.;Govind,N.J.Phys.Chem.A,2008,112:669028Cheng,M.H.;Pu,X.M.;Wong,N.B.New J.Chem.,2008,32(6): 106029Xia,Y.;Yin,D.L.;Rong,C.Y.;Xu,Q.;Yin,D.H.;Liu,S.B.J.Phys.Chem.A,2008,112:9970706。
氰乙基结构式
摘要:
1.氰乙基结构式简介
2.氰乙基结构式的化学性质
3.氰乙基结构式的应用领域
4.氰乙基结构式的安全风险及防护措施
正文:
氰乙基结构式是一种有机化合物,其化学式为C2H2N3。
在这个分子中,一个碳原子与三个氮原子相连,其中一个氮原子上连接着一个氰基(-CN)。
氰乙基结构式的形状类似于一个倒置的三角形,其中一个角为碳原子,另外两个角为氮原子,氰基则位于三角形的底部。
氰乙基结构式的化学性质使其在许多领域具有广泛的应用。
例如,在化学工业中,氰乙基结构式常用作合成中间体,用于生产染料、塑料和药物等。
此外,氰乙基结构式还可以用于电镀、金属表面处理等领域。
然而,氰乙基结构式也存在一定的安全风险。
由于氰基的化学性质非常活泼,氰乙基结构式在受热、遇酸或接触金属时,容易发生分解反应,释放出氰化氢等有毒气体。
因此,在处理氰乙基结构式时,需要采取一定的防护措施,如佩戴防护面具、手套和防护服,确保实验室通风良好等。
总之,氰乙基结构式是一种具有广泛应用的有机化合物,但同时也存在一定的安全风险。
氰乙基对几种芳胺结构和光谱的影响唐智勇1胡云楚1,*赵莹1刘述斌2,*(1中南林业科技大学应用化学研究所,长沙410004;2Research Computing Center,University of North Carolina,Chapel Hill,North Carolina 27599-3420,USA)摘要:采用量子化学中密度泛函理论(DFT)的B3LYP 方法分别用6-31G *和6-311+G *基组对苯胺、对氯苯胺和对甲苯胺及其氰乙基衍生物的几何构型进行全优化,探讨了氨基上氰乙基的引入对分子电荷转移、前线轨道能量和电子光谱等性质的影响规律.在此基础上采用含时密度泛函方法(TD -DFT)计算了分子第一激发态的电子跃迁能,得到最大吸收波长λmax .计算结果表明,氨基上氰乙基的引入,对前线分子轨道组成影响虽然小,但使得最大吸收波长红移,与实验值λmax 有较好的一致性,发现该类物质主要吸收光谱源于分子内的π→π*的电子跃迁.关键词:芳胺;氰乙基;密度泛函理论;前线轨道;电子光谱中图分类号:O641Impact of Cyanoethy Groups on Structure and Spectroscopy of a FewAromatic AminesTANG Zhi -Yong 1HU Yun -Chu 1,*ZHAO Ying 1LIU Shu -Bin 2,*(1Institute of Applied Chemistry,Central South University of Forestry &Technology,Changsha410004,P.R.China ;2Research Computing Center,University of North Carolina,Chapel Hill,North Carolina27599-3420,USA )Abstract :The impact of introducing cyanoethy groups in aromatic amines on structure,charge transfer,frontier orbital energy and composition,as well as electronic absorption spectra was investigated in this work.Theoretical studies of aniline,p -chloroaniline,p -toluidine,and cyanoethy derivatives were carried out at density functional theory (DFT)B3LYP/6-31G *and B3LYP/6-311+G *levels to obtain optimized geometrical structures.Time -dependent density functional theory was applied to calculate the first excited state electronic transition energy and maximum absorption wavelength λmax .Our results indicate that the introduction of a cyanoethy group has limited effect on the composition of frontier orbitals.The main absorption spectrum originates from the featured π→π*electronic transition.The predicted spectra agree well with the available experimental findings.Key Words :Aromatic amine;Cyanoethy;Density functional theory;Frontier orbital;Electronic spectrum[Article]物理化学学报(Wuli Huaxue Xuebao )Acta Phys.-Chim.Sin .,2009,25(4):701-706Received:October 29,2008;Revised:December 15,2008;Published on Web:February 16,2009.*Corresponding authors.Email:hucsfu@,shubin@;Tel:001-919-962-4032.2009年度湖南省研究生创新基金和2008年度中南林业科技大学研究生创新基金资助项目鬁Editorial office of Acta Physico -Chimica Sinica芳香胺的氰乙基化反应是有机合成中最基本的反应之一,其氰乙基化产物是分散染料、医药、表面活性剂的重要中间体,可广泛用于染料、抗氧化剂、抗菌剂、表面活性剂等精细化学品的合成.Allen 等[1]采用CuOAc ·H 2O 作催化剂,系统研究了10多种芳胺的单氰乙基化反应,其单氰乙基产物的收率大多小于80%,其中对氯苯胺的单氰乙基收率为78%;钟为慧等[2]采用SmI 3作催化剂研究了5种活性高的芳胺氰乙基化反应,单氰乙基化收率为59%-83%;郑立红等[3]采用CuCl 作催化剂,利用邻氯苯胺循环来合成N -氰乙基邻氯苯胺,其收率接近75%.总之,从国内外对芳胺的单氰乙基化反应的April 701Acta Phys.-Chim.Sin.,2009Vol.25研究结果来看,未有一种催化剂能使反应的转化率达到90%以上.而对于芳胺的双氰乙基化反应,有关报道却很少.仅有Braunholtz 等[4]以HAc -CuCl 催化体系反应16h 合成N ,N -二氰乙基对氯苯胺,收率只有20%.可见,要获得双氰乙基物较难.因此工业上双氰乙基产物的获得一般采用分离单、双氰乙基产物的办法.近几年来,赵莹等[5-9]开展了一系列用无水AlCl 3催化芳胺与丙烯腈进行加成反应的研究工作,合成出10多种氰乙基加成产物,收率达76%-98%,有的已经实现工业化生产并被用来合成分散染料.然而对于芳香胺的氰乙基化产物的理论研究尚未见报道.目前,应用于激发态能量计算的理论方法有多种[10],而用含时密度泛函方法(TD -DFT)[11-19]处理电子激发态问题成为当前一个非常活跃的研究领域.本研究运用Guassian 03量子化学程序包[20]中的密度泛函理论(DFT)[21-23],采用B3LYP [24,25]方法分别在6-31G *和6-311+G *水平下对合成的一系列芳香胺的氰乙基化产物进行了量子化学计算,从理论上预测氨基上氰乙基的引入对芳胺分子电荷转移、前线轨道能量和电子光谱等性质的影响规律,探讨此类分子中第一激发态电子跃迁的本质,并通过实验验证了几种芳胺及氰乙基芳胺光谱规律,从而为设计新型染料结构提供理论依据.1计算模型和方法运用Guassian 03量子化学程序包[20]中的密度泛函理论[21-23],首先在B3LYP/6-31G *和B3LYP/6-311+G *水平上,对几种芳香胺及其氰乙基衍生物(图1)进行全自由度几何优化,并做频率分析,频率分析结果均无虚频,表明选用的目标化合物的优化结果确实为其稳定构型.以基态的优化构型为基础,应用含时密度泛函理论TD -DFT [11-19],分别在相同理论水平(B3LYP/6-31G *和B3LYP/6-311+G *)上计算化合物的电子吸收光谱,并与实验结果进行比较,计算中所采用的收敛精度为程序设定值.将6-31G *和6-311+G *两个基组得到的结果进行比较,发现几何构型、净电荷布居和前线轨道数据相差甚微,不影响全文对结构的分析,而在最大吸收波长方面有较大的差别,所以,结果与讨论中的前3节只列出了6-31G *水平下的数据,在第4节中讨论了两种水平下的最大吸收波长.2结果与讨论2.1几何构型表1给出了在6-31G *水平下9种芳胺分子的部分优化键长、键角和二面角.从表1可以看出,L (6,7)的C —Cl 键长均小于饱和卤代烷烃中C —Cl 键长(0.177nm)[26],原因是由于氯原子的p 轨道与苯环上π键有部分重叠,因此,C —Cl 键之间也有部分双键的性质,使得C —Cl 键长变短;L (6,7)的C —C 键长均介于C —C 单键(0.154nm)[26]和C 襒C 双键(0.134nm)[26]之间,表明具有一定的双键性质,引入氰乙基后,对L (6,7)的C —Cl 和C —C 几乎没有影响,原因是氰乙基处于苯胺分子的对位,距离较远;777991010R1=H,CH 3,Cl;R2=H,CH 2CH 2CN;R3=H,CH 2CH 2CN图1芳香胺及其衍生物的分子结构模型和原子编号Fig.1Structure model and atomic number ofaromatic amine and its derivativesL :bond length in nm;D :dihedral angle in degree;X:CH 2CH 2CN;optimized at B3LYP/6-31G *level表1芳香胺及其衍生物的几何构型及其优化参数Table 1Optimized geometries and structural characteristics of aromatic amine and its derivativesCompoundA1A2A3B1B2B3E1E2E3R1H H H Cl Cl Cl CH 3CH 3CH 3R2H H X H H X H H X R3H X X H X X H X X L (6,7)0.10870.10860.10860.17610.17610.17580.15110.15110.1511L (3,8)0.14460.14020.14050.14430.14000.14020.14020.14010.1406L (8,9)0.10200.10150.14500.10190.10140.14560.10130.10130.1450L (8,10)0.10200.14500.14500.10190.14520.14510.10130.14480.1450D (4,5,6,7)-180.00179.72-179.68180.00179.69179.71-178.59179.68178.45D (1,2,3,8)180.00-177.91178.72180.00-177.88-179.06-177.22178.20178.38D (2,3,8,9)-58.04-27.043.43-58.28-26.816.11-155.9425.042.16D (2,3,8,10)58.08-164.48179.8258.41-165.80177.95-26.80165.59178.57702No.4唐智勇等:氰乙基对几种芳胺结构和光谱的影响calculated at B3LYP/6-31G *levelL (3,8),L (8,9)和L (8,10)的C —N 键长均介于C —N 单键(0.147nm)[26]和C 襒N 双键(0.127nm)[26]之间,表明也具有一定的双键性质,与苯环形成共轭.几何构型全优化后,系列苯胺分子的主要骨架其二面角均接近于-180°或180°,氨基上两个氢原子都被氰乙基取代后,与氨基相连氰乙基碳的二面角接近于0°和180°,说明氰乙基引入后,导致氨基构型发生扭转,分子平面延伸.苯胺、对氯苯胺和对甲苯胺分子结构中主要原子和原子团基本能保持在同一平面上.故这些原子和苯环一起构成了大的共轭体系.2.2净电荷布居表2给出了在6-31G *水平下9种芳胺分子部分原子的净电荷布居,从表2可以看出:氰乙基取代后的几种苯胺分子发生了不同程度的电荷转移,与其直接键连的原子的电荷分布变化较大,离取代基越远,原子上的电荷分布变化越小.化合物B1由于Cl 的吸电子诱导效应大于p -π共轭效应,使得C(6)上净电荷明显降低;而化合物E1苯环上所有C 的净电荷都明显升高,尤其是C(6),所带的电荷由负变正,说明甲基主要以p -π共轭效应为主,电子云向相邻碳转移.氨基与苯环形成p -π共轭,由于氰乙基的吸电子诱导效应,引入后使得氨基N 原子上负的净电荷明显降低,程度顺序为双氰乙基>单氰乙基;C(3)上正的净电荷明显增加,程度顺序为双氰乙基>单氰乙基,对位上有吸电基Cl 或供电基CH 3几乎无影响.化合物的反应活性与分子中原子所带净电荷的多少有关,电荷集中的原子就是分子反应的活性中心.原子的正电荷越多,受亲核试剂进攻的可能性越大;反之,原子的负电荷越多,其受亲电试剂进攻的可能性越大.因此,从表2中可以看出,负电荷主要集中在氨基N(8)上,可以预测N(8)是亲电反应的作用点,有利于亲电试剂丙烯腈进攻.正是由于N 原子上负电荷远大于苯环上各碳原子,才使得几种苯胺与丙烯腈反应,生成的是N -氰乙基苯胺和N ,N -二氰乙基苯胺,而不是苯环上氰乙基取代苯胺,这点在文献[1-5]中已得到证实.2.3前线轨道能量量子化学的前线轨道理论认为,分子参与反应则分子轨道要发生变化,优先起作用的是前线轨道,即分子中的最高占有轨道(HOMO)和最低空轨道(LUMO).对分子轨道的分析可以为我们提供很多有关分子的信息,比如电子的结合位置及分子的化学反应性能[27-29],此外,系列苯胺分子的前线轨道能量与光电子能谱密切相关,而前线轨道能量差与紫外-可见光谱性质密切相关.表3给出了在6-31G *水平下9种芳胺分子的表3芳香胺及其衍生物前线轨道能级(a.u.)Table 3Frontier molecular orbital energy levels (a.u.)of aromatic amine and its derivatives表2芳香胺及其衍生物部分原子的净电荷布居(e )Table 2Net charge populations (e )of aromatic amine and its derivativesCompoundA1A2A3B1B2B3E1E2E3C(1)-0.138-0.134-0.137-0.139-0.135-0.140-0.188-0.184-0.192C(2)-0.156-0.188-0.197-0.152-0.183-0.189-0.170-0.184-0.195C(3)0.2060.3500.3570.2100.3570.3620.3020.3490.354C(4)-0.125-0.183-0.197-0.121-0.177-0.190-0.170-0.179-0.195C(5)-0.140-0.138-0.137-0.140-0.141-0.140-0.188-0.196-0.190C(6)-0.126-0.136-0.133-0.061-0.071-0.0680.1750.1790.182C(7)-0.529-0.531-0.531Cl(7)-0.029-0.034-0.026N(8)-0.778-0.668-0.489-0.779-0.670-0.490-0.787-0.654-0.491C(9)-0.157-0.158-0.156C(10)-0.112-0.157-0.111-0.161-0.102-0.156Compound A1A2A3B1B2B3E1E2E3HOMO -0.2404-0.2034-0.2147-0.2414-0.2126-0.2171-0.1922-0.1973-0.2086LUMO 0.00130.0025-0.0148-0.0139-0.0159-0.02630.00980.0035-0.0137ΔE0.24170.20590.19990.22750.19670.19080.20200.20070.1949calculated at B3LYP/6-31G *level703Acta Phys.-Chim.Sin.,2009Vol.25前线轨道能级.从表3可以看出,对位Cl 取代使分子的前线轨道能降低,对位CH 3取代使分子的前线轨道能量升高.取代基的引入使分子能隙变小,供电子基CH 3对能隙的减小更为明显,氰乙基的引入及数目增加对减小能隙起促进作用.能隙大小顺序为苯胺>单氰乙基苯胺>双氰乙基苯胺.ΔE 随氰乙基的引入逐渐变小,表明吸收光谱的最大吸收峰可能会出现红移现象.计算得到的能隙大小顺序,预示着系列苯胺分子电子光谱最大吸收波长的大小顺序.从HOMO 轨道和LUMO 轨道的能量上看,能量变化较大,它们是反应的主要参与轨道.为了较全面地利用轨道理论来解释分子反应活性,画出了几种芳香胺及其氰乙基衍生物的最高占有轨道(HOMO)和最低空轨道(LUMO)的空间分布图(图2).分别将化合物分子中的所有原子的轨道组合为分子轨道系数的平方和,归一化后,得到各原子在前线分子轨道及附近分子轨道中的贡献(表4).图2给出了在6-31G *水平下9种芳胺分子的前沿分子轨道图形,从中可以看出,9种芳香胺的HOMO 和LUMO 轨道都是π分子轨道.表4给出了在6-31G *水平下9种芳胺分子HOMO 和LUMO 中主要原子的贡献百分率(为便于比较,小于1.5的数值忽略不计),从表4可以看出,化合物B1的表4前线轨道中各主要原子的贡献百分率(%)Table 4Main atomic contributive percents (%)of frontier orbital for molecular orbital图2芳香胺及其衍生物的前沿分子轨道Fig.2Frontier molecular orbitals of aromatic amine and its derivativesMain atomC(1)C(2)C(3)C(4)C(5)C(6)Cl/C(7)N(8)C(9)C(10)A1HOMO 17.82 6.6728.0310.17 4.9327.75 1.78LUMO 5.6032.6812.60 4.5731.8011.34-A2HOMO 5.6210.899.589.71 4.4617.8637.59 3.07LUMO 20.7727.72-19.7527.50---A3HOMO 6.9110.437.0410.43 2.4316.7137.29--LUMO 20.4118.46-18.8920.42-- 3.303.40B1HOMO 14.46 4.6819.53 4.99 6.1918.9526.70 1.64LUMO 6.9930.778.69 6.6129.968.76--B2HOMO 6.299.059.218.44 3.4915.9112.1031.35-LUMO 21.6227.81-19.8927.61----B3HOMO 8.119.11 6.858.57 2.3915.1411.1331.51--LUMO 23.7222.60-21.8723.72-----E1HOMO 7.2110.4312.3610.44 2.0619.77-31.10LUMO 22.3121.81-21.8221.80---E2HOMO 7.469.339.9410.28 2.1216.85-34.39-LUMO 25.9225.35-22.5223.94----E3HOMO 8.2010.449.629.85 1.8317.11-37.39--LUMO22.7322.19-20.2625.35-----calculated at B3LYP/6-31G *level704No.4唐智勇等:氰乙基对几种芳胺结构和光谱的影响λmax in nm;a)excitation energies at the TD-DFT/6-31G*level,b)oscillator strengths at the TD-DFT/6-31G*level,c)excited state,d)calculated at the TD-DFT/6-31G*level;e)calculated at the TD-DFT/6-311+G*level,f)experimental data were determined in the solvent of ethanol.HOMO电子云主要集中在苯环和对位Cl(7)上,其中Cl(7)占26.70%,与之相连的C(6)占18.95%,而N(8)上电子分布几乎为0,其原因是,虽然Cl(7)和N(8)都与苯环形成了p-π共轭,但是Cl的电负性大于N,吸电子诱导效应大于共轭效应,所以电子云主要集中于C(6)和Cl(7)上;化合物E1的HOMO电子云主要集中在苯环和N(8)上,其中N(8)占31.10%,而对位CH3上接近于0,这是因为CH3是推电子基,使得电子云主要离域在N(8)周围;化合物A2、A3、B2、B3、E2和E3的HOMO电子云主要分布在N(8)和苯环上,其中N(8)都占31%以上,主要是N 的电负性大于C和氰乙基的吸电子诱导效应.9个化合物的LUMO轨道电子云都分散在苯环上.从图2中能更加清晰地看出其前线轨道组成特征.由此可以预测对氯苯胺难与丙烯腈发生亲电取代反应,而对甲苯胺易与丙烯腈发生亲电取代反应.通过实验也证实了这点,在无水AlCl3催化下,前者回流反应24h,产率只有85%左右[6];后者回流反应12h,产率有93%以上[5].2.4电子吸收光谱近年来,TD-DFT方法已越来越多地被应用于有机化合物光谱性质的研究.我们采用UV-1100紫外-可见分光光度计以无水乙醇为溶剂,分别测定了苯胺、对氯苯胺和对甲苯胺及其氰乙基衍生物9种化合物的紫外-可见光谱,它们的最大吸收波长列于表5中.表5给出了几种芳胺及其氰乙基衍生物在6-31G*和6-311+G*水平下的最大吸收波长.从表5中可以看到,系列苯胺分子的最大吸收波长计算值和实验值之间有一定差异,可能在于程序计算是按理想气态分子处理的,而实验值是以无水乙醇为溶剂获得的,但仍有着较好的一致性.除化合物A1和B1外, 6-31G*水平下λmax大都比实验λmax小约38nm,而6-311+G*水平下λmax基本上比实验λmax小约24nm,由此可知,6-311+G*基组在分析小分子芳胺光谱性质上比6-31G*基组相对准确些,从系列苯胺分子的最大吸收波长的跃迁能量顺序来看,与表3的能隙顺序一致.系列苯胺分子的最大吸收波长均位于近紫外区,λmax计算值按—NH2、—NHCH2CH2CN、—N(CH2CH2CN)2顺序红移,这与实验值一致.计算结果较好地反映实验的变化规律.3结论对自行合成的系列苯胺分子进行了量子化学计算,从理论上预测了氨基上—CH2CH2CN的引入对分子电荷转移、前线分子轨道能量和电子吸收光谱等性质的影响规律.通过理论计算并与实验结果比较发现,采用DFT-B3LYP方法研究简单苯胺分子及其氰乙基衍生物可以得到较合理的几何构型和电子吸收光谱结果.通过对分子中主要原子净电荷的分析,揭示了诱导效应和共轭效应影响9种苯胺分子电荷分布,由主要原子所带电荷多少知道几种苯胺分子的反应活性中心以及与亲电试剂丙烯腈反应的难易程度.通过对前线轨道的分析,认为9种苯胺分子的HOMO和LUMO均为π分子轨道,其主要由氨基和苯环所贡献.分子轨道研究表明,此类化合物最大吸收波长是分子吸收能量之后电子从HOMO向LUMO的跃迁,即电子从共轭体系中给电子体跃迁到受电子体引起的.取代基的引入,均导致最大吸收波长红移,吸电子基引起的红移程度大于供电子基,理论计算与实验结果的规律基本一致.根据化学结构和电子吸收光谱可以预测这一系列苯胺分子的紫外吸收区,从而为染料的合成,如测定其敏化区的位置提供帮助.了解芳胺类分子电子结构和光谱性质是理解其作为光敏材料、染料中间体和进行分子设计的起点.故在DFT框架内表5芳香胺及其衍生物的λmax计算值与实验值的比较Table5Comparison of calculated and experimental data ofλmax for aromatic amine and its derivatives Compound A1A2A3B1B2B3E1E2E3E a 5.4704 5.3312 4.7244 5.2713 4.6728 4.5231 4.8079 4.7698 4.6257f b0.00210.02600.03660.00730.03200.03360.03280.03000.0376 Characterπ→π*π→π*π→π*π→π*π→π*π→π*π→π*π→π*π→π* Transition c25→2639→4053→5429→3043→4457→5833→3447→4861→62λmax(calc.)d226.64252.56262.44235.21265.33274.11257.88259.94268.03λmax(calc.)e232.42267.09271.07242.11278.18284.26271.34273.96278.52λmax(expt.)f278.5288.5295.5300.0306.0310.0293.0298.0301.0705Acta Phys.-Chim.Sin.,2009Vol.25进行结构与性质的研究,尤其是应用含时密度泛函理论对此类芳胺中间体作单电子跃迁的计算,将为以后改构设计分子提供参考依据.References1Allen,.Chem.,1957,22:12132Zhong,W.H.;Zhang,.Chem.,2000,20(5): 747[钟为慧,张永敏.有机化学,2000,20(5):747]3Zheng,L.H.;Wang,X.H.;Zhao,C.Y.Dyestuff Industry,2001,4: 26[郑立红,王宪花,赵从伊.染料工业,2001,4:26]4Braunholtz,J.T.;Mann,F.G.J.Chem.Soc.,1953:18175Zhao,Y.;Tan,X.Y.;Yang,Z.;Li,.Chem.,2005, 25(11):1469[赵莹,谭晓艳,杨志,李姣娟.有机化学,2005,25(11):1469]6Zhao,Y.;Ye,C.C.;Li,J.J.;Yang,Z.Chemistry,2005,69(6):438 [赵莹,叶翠层,李姣娟,杨志.化学通报,2005,69(6):438] 7Gong,J.L.;Li,J.J.;Zhao,Y.Dyestuffs and Coloration,2008,45(1):44[龚建良,李姣娟,赵莹.染料与染色,2008,45(1):44] 8Zhao,Y.;Ye,C.C.Chemical Reagents,2006,28(5):81[赵莹,叶翠层.化学试剂,2006,28(5):81]9Zhao,Y.;Tan,X.Y.;Li,J.J.;Yang,Z.Chin.J.Syn.Chem.,2006, 14(3):297[赵莹,谭晓燕,李姣娟,杨志.合成化学,2006, 14(3):297]10Liu,S.F.;Seward,C.;Aziz,H.;Hu,N.X.;Popovic,Z.;Wang,S.anometal.,2000,19(26):570911Belletete,M.;Durocher,G.;Hamel,S.;Cote,M.;Wakim,S.;Leclerc,M.J.Chem.Phys.,2005,122(10):4303112Fan,J.X.;Ren,A.M.;Feng,J.K.;Bo,D.S.Chem.J.Chin.Univ., 2006,27(6):1091[范建训,任爱民,封继康,薄东生.高等学校化学学报,2006,27(6):1091]13Xu,H.;Yang,B.;He,F.;Xie,Z.Q.;Tian,L.L.;Liu,X.D.;Yu,J.S.;Ma,Y.G.Chem.J.Chin.Univ.,2006,27(3):510[许海,杨兵,何凤,解增旗,田雷蕾,刘晓冬,于景生,马於光.高等学校化学学报,2006,27(3):510]14Li,H.X.;Xiao,T.Chem.J.Chin.Univ.,2007,28(4):747 [李会学,萧泰.高等学校化学学报,2007,28(4):747]15Liu,J.N.;Chen,Z.R.;Yuan,S.F.Acta Phys.-Chim.Sin.,2005, 21(4):402[刘军娜,陈志荣,袁慎峰.物理化学学报,2005,21(4):402]16Liang,X.Q.;Pu,X.M.;Shu,Y.J.;Tian,A.M.Acta.Chim.Sin., 2006,64(20):2057[梁晓琴,蒲雪梅,舒远杰,田安民.化学学报,2006,64(20):2057]17Chen,Q.W.;Wang,L.Y.;Zhai,G.H.;Wen,Z.Y.;Zhang,Z.X.Acta Chim.Sin.,2005,63(1):39[陈沁闻,王兰英,翟高红,文振翼,张祖训.化学学报,2005,63(1):39]18Xue,Y.S.;Gong,X.D.;Xiao,H.M.;Tian,H.Acta Chim.Sin., 2004,62(10):963[薛运生,贡雪东,肖鹤鸣,田禾.化学学报, 2004,62(10):963]19Runge,E.;Gross,E.K.U.Phys.Rev.Lett.,1984,52:99720Frish,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian03, Revision B.02.Pittsburgh PA:Gaussian,Inc.,200321Marques,M.A.L.;Gross,E.K.U.Annu.Rev.Phys.Chem.,2004, 55:42722Dreuw,A.;Head-Gordon,M.Chem.Rev.,2006,105:4009.23Huang,Y.;Zhong,A.G.;Rong,C.Y.;Xiao,X.M.;Liu,S.B.J.Phys.Chem.A,2008,112:30524Becke,A.D.J.Chem.Phys.,1992,97:917325Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B,1988,37:78526Tao,S.X.;Zhao,J.W.Advanced organic chemistry.Beijing: People′s Education Press,1981:16[陶慎熹,赵景文.高等有机化学.北京:人民教育出版社,1981:16]27Liu,S.B.;Govind,N.J.Phys.Chem.A,2008,112:669028Cheng,M.H.;Pu,X.M.;Wong,N.B.New J.Chem.,2008,32(6): 106029Xia,Y.;Yin,D.L.;Rong,C.Y.;Xu,Q.;Yin,D.H.;Liu,S.B.J.Phys.Chem.A,2008,112:9970706。