新版GMP-无菌药品附录
- 格式:ppt
- 大小:900.50 KB
- 文档页数:50

附录1:无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括注射剂、眼用制剂、无菌软膏剂、无菌混悬剂等。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第三条悬浮粒子、浮游菌、沉降菌和表面微生物等测试方法应按照相关标准执行。
第二章原则第四条无菌药品的生产须满足其质量和预定用途的要求,应最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验。
第五条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第六条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,如采用机械连续传输物料时,应采用正压气流保护并监测压差。
物料准备、产品配制和灌装或分装等操作必须在洁净区内分区(室)进行。
第七条应按所需环境的特点确定无菌药品洁净生产区的级别。
每一步生产操作的环境都应达到适当的动态洁净度标准,以尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级高风险操作区,如:灌装区、放置胶塞桶、敞口安瓿瓶、敞口西林瓶的区域及无菌装配或连接操作的区域。
通常用层流操作台(罩)来维持该区的环境状态。
层流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应有数据证明层流的状态并须验证。
在密闭的隔离操作器或手套箱内,可使用单向流或较低的风速。
B级指无菌配制和灌装等高风险操作A级区所处的背景区域。
C级和D级指生产无菌药品过程中重要程度较低的洁净操作区。
以上各级别空气悬浮粒子的标准规定如下表:注:(1) 为了确定A级区的级别,每个采样点的采样量不得少于1m3。

药品生产质量管理规范(新版GMP)附录:无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
以上各级别空气悬浮粒子的标准规定如下表:新版GMP附录无菌药品注:(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。


无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查工程的制剂和原料药,包括无菌制剂和无菌原料药。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
以上各级别空气悬浮粒子的标准规定如下表:注:(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方M。

新版GMP附录一、无菌制剂二、原料药三、生物制品四、血液制品五、中药制剂一、无菌制剂第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。

《药品生产质量管理规范(2010年修订)》发布2011年02月12日发布历经5年修订、两次公开征求意见的《药品生产质量管理规范(2010年修订)》(以下简称新版药品GMP)今天对外发布,将于2011年3月1日起施行。
《药品生产质量管理规范》(以下简称药品GMP)是药品生产和质量管理的基本准则。
我国自1988年第一次颁布药品GMP至今已有20多年,其间经历1992年和1998年两次修订,截至2004年6月30日,实现了所有原料药和制剂均在符合药品GMP的条件下生产的目标。
新版药品GMP共14章、313条,相对于1998年修订的药品GMP,篇幅大量增加。
新版药品GMP吸收国际先进经验,结合我国国情,按照“软件硬件并重”的原则,贯彻质量风险管理和药品生产全过程管理的理念,更加注重科学性,强调指导性和可操作性,达到了与世界卫生组织药品GMP的一致性。
药品GMP的修订是药监部门贯彻落实科学发展观和医疗卫生体制改革要求,进一步关注民生、全力保障公众用药安全的又一重大举措,它的实施将进一步有利于从源头上把好药品质量安全关。
1998年修订的药品GMP的实施,在提升我国药品质量、确保公众用药安全方面发挥了重要的作用,取得了良好的社会效益和经济效益。
随着经济的发展和社会的进步,世界卫生组织及欧美等国家和地区药品GMP的技术标准得到很大的提升,新的理念和要求不断更新和涌现,我国现行药品GMP需要与时俱进,以适应国际药品GMP发展趋势,也是药品安全自身的要求。
我国现有药品生产企业在整体上呈现多、小、散、低的格局,生产集中度较低,自主创新能力不足。
实施新版药品GMP,是顺应国家战略性新兴产业发展和转变经济发展方式的要求。
有利于促进医药行业资源向优势企业集中,淘汰落后生产力;有利于调整医药经济结构,以促进产业升级;有利于培育具有国际竞争力的企业,加快医药产品进入国际市场。
新版药品GMP修订的主要特点:一是加强了药品生产质量管理体系建设,大幅提高对企业质量管理软件方面的要求。

附录1 无菌产品生产文件日期:2020-02-20目录•文件结构图• 1 范围• 2 原则• 3 药品质量体系(PQS)• 4 厂房• 5 设备• 6 公共设施•7 人员•8 生产和具体技术•9 活性微粒及非活性微粒的环境监测和工艺监测•10 质量控制(QC)•11.术语1 范围无菌产品的生产涵盖多种无菌产品类型(原料药,无菌辅料,内包装材料和成品制剂),包装规格(单剂量到多剂量),工艺(从高度自动化系统到手动工艺)和技术(如生物技术,传统小分子生产和密闭系统)。
本附录提供了运用质量风险管理(QRM)原则的所有无菌产品生产应施用的一般性指导,以确保最终产品中无微生物、微粒和热原污染。
QRM缩写适用于本文件全文,不会在具体段落中加以说明。
在列出具体限度或频率时,这些限度或频率应视为最低要求。
这些陈述是由于监管历史经验,即曾出现这些问题并影响了患者的安全。
本附录的目的是为无菌产品的生产提供指导。
然而,一些原则和指导,例如污染控制策略、厂房设计、洁净室分类、确认、监测和人员更衣,可用于支持其它非无菌、但有必要控制和减少微生物、微粒和热原污染的产品(例如某些液体、乳膏、软膏和低生物负荷的生物中间体)的生产。
如果生产商选择将本指南应用于非无菌产品,生产商应清楚地记录已施用了哪些原则,并应证明符合这些原则。
2 原则2.1 无菌产品的生产应符合特殊要求,以尽量降低微生物、微粒和热原污染的风险。
应考虑以下关键领域:i.应按照药品生产质量管理规范(GMP)指南的相关章节优化、确认和验证设施、设备和工艺设计。
应考虑使用适当的技术(例如,限制进入隔离系统(RABS),隔离器,自动系统,快速微生物检测和监测系统),以增强对产品的保护,防止潜在的外来微粒和微生物污染源(例如人员、物料和周围环境),并帮助快速检测环境和产品中的潜在污染物。
ii.人员应具备合适的资质和经验,培训和态度,尤其是生产、包装和发运过程中无菌产品保护所涉及的原则。


《药品生产质量管理规范(2010年修订)》1发布232011年02月12日发布456历经5年修订、两次公开征求意见的《药品生产质7量管理规范(2010年修订)》(以下简称新版药品GMP)今8天对外发布,将于2011年3月1日起施行。
9《药品生产质量管理规范》(以下简称药品GMP)是药10品生产和质量管理的基本准则。
我国自1988年第一次颁布11药品GMP至今已有20多年,其间经历1992年和1998年两12次修订,截至2004年6月30日,实现了所有原料药和制13剂均在符合药品GMP的条件下生产的目标。
新版药品GMP14共14章、313条,相对于1998年修订的药品GMP,篇幅大15量增加。
新版药品GMP吸收国际先进经验,结合我国国情,16按照“软件硬件并重”的原则,贯彻质量风险管理和药品17生产全过程管理的理念,更加注重科学性,强调指导性和18可操作性,达到了与世界卫生组织药品GMP的一致性。
19药品GMP的修订是药监部门贯彻落实科学发展观和医疗卫生体制改革要求,进一步关注民生、全力保障公众2021用药安全的又一重大举措,它的实施将进一步有利于从源22头上把好药品质量安全关。
1998年修订的药品GMP的实施,23在提升我国药品质量、确保公众用药安全方面发挥了重要24的作用,取得了良好的社会效益和经济效益。
随着经济的25发展和社会的进步,世界卫生组织及欧美等国家和地区药26品GMP的技术标准得到很大的提升,新的理念和要求不断27更新和涌现,我国现行药品GMP需要与时俱进,以适应国28际药品GMP发展趋势,也是药品安全自身的要求。
29我国现有药品生产企业在整体上呈现多、小、散、低的格局,生产集中度较低,自主创新能力不足。
实施新3031版药品GMP,是顺应国家战略性新兴产业发展和转变经济发32展方式的要求。
有利于促进医药行业资源向优势企业集中,33淘汰落后生产力;有利于调整医药经济结构,以促进产业34升级;有利于培育具有国际竞争力的企业,加快医药产品35进入国际市场。

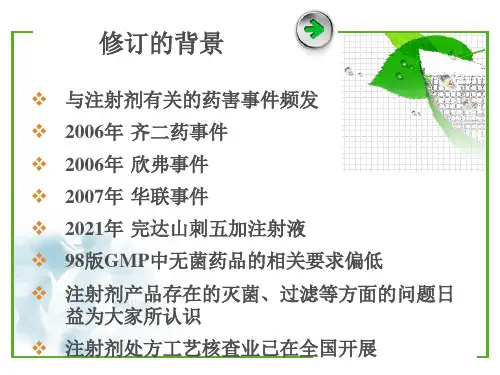
新版gmp附录无菌药品完全解读GMP(Good Manufacturing Practice,良好生产规范)是指药品生产中所需的标准和过程,用于确保药品的质量和安全性。
在GMP中,附录无菌药品是指在无菌条件下制备、包装和储存的药品。
下面是对新版GMP附录无菌药品的完全解读:1. 无菌条件:药品的生产过程必须在严格的无菌条件下进行。
这包括无菌区域的建设与维护,严格的人员培训和操作规程,以及使用有效的无菌技术和设备。
2. 无菌验证:对无菌药品生产过程中的无菌性进行验证是必要的。
这可以通过使用微生物培养基和培养方法,以及对生产设备和包装容器进行微生物测试来实现。
3. 生物污染风险评估:对可能存在的生物污染源进行系统评估,制定相应的风险控制措施。
这包括对原辅材料、生产区域和人员的风险评估,以及制定相应的控制策略。
4. 标识和包装:对无菌药品的标识和包装要求严格。
这包括药品容器和包装材料的无菌性检验,以及对包装过程和包装材料的有效性验证。
5. 清洁和消毒:无菌药品生产设备和无菌区域的清洁和消毒非常重要。
这包括合适的清洁剂和消毒剂的使用,以及相应的清洁和消毒程序的制定和执行。
6. 培训和认证:对参与无菌药品生产的人员进行培训和认证是必要的。
这包括无菌操作技术的培训,正确使用无菌设备的培训,以及对无菌操作流程的监督和检查。
7. 环境监测:对无菌区域和生产设备的环境进行定期监测是必要的。
这包括空气微生物和表面菌落的定期检测,以及对环境监测结果的分析和处理。
总的来说,新版GMP附录无菌药品的要求更加严格和详细,要求生产企业建立、实施和维护科学的无菌药品生产过程,确保药品的质量、安全和有效性。
新版GMP附录GMP一、无菌制剂二、原料药三、生物制品四、血液制品五、中药制剂一、无菌制剂第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
《药品生产质量管理规范(2010年修订)》发布2011年02月12日发布历经5年修订、两次公开征求意见的《药品生产质量管理规范(2010年修订)》(以下简称新版药品GMP)今天对外发布,将于2011年3月1日起施行。
《药品生产质量管理规范》(以下简称药品GMP)是药品生产和质量管理的基本准则。
我国自1988年第一次颁布药品GMP至今已有20多年,其间经历1992年和1998年两次修订,截至2004年6月30日,实现了所有原料药和制剂均在符合药品GMP的条件下生产的目标。
新版药品GMP共14章、313条,相对于1998年修订的药品GMP,篇幅大量增加。
新版药品GMP吸收国际先进经验,结合我国国情,按照“软件硬件并重”的原则,贯彻质量风险管理和药品生产全过程管理的理念,更加注重科学性,强调指导性和可操作性,达到了与世界卫生组织药品GMP的一致性。
药品GMP的修订是药监部门贯彻落实科学发展观和医疗卫生体制改革要求,进一步关注民生、全力保障公众用药安全的又一重大举措,它的实施将进一步有利于从源头上把好药品质量安全关。
1998年修订的药品GMP的实施,在提升我国药品质量、确保公众用药安全方面发挥了重要的作用,取得了良好的社会效益和经济效益。
随着经济的发展和社会的进步,世界卫生组织及欧美等国家和地区药品GMP的技术标准得到很大的提升,新的理念和要求不断更新和涌现,我国现行药品GMP需要与时俱进,以适应国际药品GMP发展趋势,也是药品安全自身的要求。
我国现有药品生产企业在整体上呈现多、小、散、低的格局,生产集中度较低,自主创新能力不足。
实施新版药品GMP,是顺应国家战略性新兴产业发展和转变经济发展方式的要求。
有利于促进医药行业资源向优势企业集中,淘汰落后生产力;有利于调整医药经济结构,以促进产业升级;有利于培育具有国际竞争力的企业,加快医药产品进入国际市场。
新版药品GMP修订的主要特点:一是加强了药品生产质量管理体系建设,大幅提高对企业质量管理软件方面的要求。