CRISPR实验流程
- 格式:pdf
- 大小:195.01 KB
- 文档页数:7

crisprcas9基因编辑技术流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!CRISPR-Cas9 基因编辑技术流程一、准备工作阶段。

CRISPR基因编辑步骤详解基因编辑是一种通过修改DNA序列来改变有机体基因组的技术,CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)基因编辑技术则是近年来最受关注的一种基因编辑方法。
CRISPR技术不仅简单高效,还可以应用于多种生物体,包括人类,其广泛的应用前景使其成为生命科学领域的重要工具。
本文将详细介绍CRISPR基因编辑的步骤。
CRISPR基因编辑主要由以下几个步骤组成:选择目标基因,设计寡核苷酸,构建脱靶检测系统,编辑基因组,确认编辑效果。
首先,选择目标基因是进行基因编辑的关键步骤之一。
根据研究的需求和目标,科学家们需要仔细选择他们希望编辑的特定基因。
选择目标基因时,需要考虑基因功能和可能的突变对生物体的影响,以及基因的可编辑性等因素。
接下来,设计寡核苷酸是CRISPR基因编辑的重要步骤。
寡核苷酸通常有两种类型:RNA寡核苷酸(sgRNA)和单链DNA寡核苷酸(ssODN)。
sgRNA的作用是指导Cas9蛋白定位到目标基因上,而ssODN则是用于修复基因序列的片段。
因此,设计合适的寡核苷酸是确保编辑效果的关键。
构建脱靶检测系统是CRISPR基因编辑过程中一个重要的质控步骤。
由于CRISPR技术存在一定程度的脱靶效应,可能会对非目标基因进行修改,因此需要设计合适的方法来检测和排除这些脱靶效应。
常用的脱靶检测方法包括PCR扩增和DNA测序等。
编辑基因组是CRISPR技术的核心步骤。
一旦确定了目标基因和寡核苷酸的设计,科学家们就可以使用Cas9蛋白与sgRNA形成复合物,这个复合物会与目标基因的DNA序列配对,并引导Cas9蛋白在目标位点上产生双链切割。
这种双链切割会触发细胞内自然修复机制,从而导致基因组的编辑。
最后,确认编辑效果是CRISPR基因编辑中不可或缺的一步。
为了确定编辑效果,科学家们通常会使用一系列技术和方法来检测是否成功编辑了目标基因。

CRISPRCas9基因编辑操作步骤及详细说明实验材料与方法一、细胞培养人宫颈癌细胞 HeLa,常规培养使用含 10% FBS 的 DMEM 培养基 ( 含 1.5 mg/L-Glutamine,100 U/mL Penicillin,100 μg/mL Streptomycin) 中,37ºC 5% CO2 饱和湿度培养箱中培养。
二、基因信息及双 gRNA 设计基因信息及分析1.hsa-mir-152 基因信息:pubmed2.hsa-mir-152 基因位于蛋白编码基因 COPZ2 内含子内,敲除hsa-mir-152 基因不会影响该蛋白编码3.hsa-mir-152 precursor 序列(87 bp):TGTCCCCCCCGGCCCAGGTTCTGTGATACACTCCGACTCGGGCTCTGGAGCAGTCAGTGCATGACAGAACTTGGGCCCGGAAGGACC双 gRNA 设计使用在线 gRNA 设计软件在 hsa-mir-152 precursor 基因组序列两侧设计双 gRNA注:dgRNA 即为双 gRNA.三、慢病毒侵染实验材料及试剂DMEM 培养基 + 10% FBSD-Hank’s SolutionTrypsin-EDTA Solution96 孔板24 孔板Lentivirus- 病毒液(GenePharma)步骤靶细胞侵染实验1.靶细胞铺板:24-well,加入2.5×105 cells/well(根据细胞种类调整),0.5 mL 完全培养基,37℃,5% CO2 过夜;2.稀释病毒:稀释液(靶细胞维持液培养基)400 μL + 终浓度 5 μg/mL Polybrene,将慢病毒原液按 1:9 加入到稀释液中;3.移去 Step1 中细胞培养液,加入 Step2 稀释后的病毒液,同时建立对照(blank、negative),37℃,5% CO2 过夜;4.12~24 小时移去细胞侵染后的病毒液,加入 0.5 mL 完全培养基,37℃,5% CO2 过夜;5.根据细胞状态和类型,如果必要分出 1/3~1/5,加入0.5 mL 完全培养基,继续培养 24~48 小时,荧光倒置显微镜下观察结果。

基因编辑技术的CRISPR系统使用方法与注意事项基因编辑技术的出现带来了革命性的突破,使人们能够直接改变生物体的基因组。
CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)系统是一种常用的基因编辑工具,其高效性和精确性使其成为科学家们的首选。
首先,使用CRISPR系统进行基因编辑需要明确的步骤与注意事项。
第一步是设计sgRNA(single-guide RNA)。
sgRNA是CRISPR系统中的关键组成部分,它能够指导Cas9酶准确地定位到目标基因上。
设计sgRNA时,应选择与目标基因相匹配的区域,并确保不会与其它基因产生非特异性的配对。
此外,sgRNA还需要具有适当的长度、稳定的结构和高活性。
第二步是构建CRISPR-Cas9复合物。
CRISPR-Cas9复合物由Cas9酶和sgRNA组成。
Cas9酶具有切割DNA的能力,而sgRNA能够指导Cas9酶准确地定位到基因组中的特定位置。
构建CRISPR-Cas9复合物时,需确保Cas9酶与sgRNA的浓度与比例适当,以确保有效的基因编辑。
此外,还需注意保持实验条件的一致性,避免干扰实验结果。
第三步是转染CRISPR-Cas9复合物到目标细胞中。
转染是将外源DNA或RNA导入目标细胞中的过程。
目前常用的转染方法有化学转染、电穿孔转染和病毒载体转染等。
选择合适的转染方法需要考虑目标细胞类型、实验目的和转染效率等因素。
第四步是筛选和验证编辑后的细胞或生物体。
基因编辑后,需要对细胞或生物体进行筛选和验证,以确认目标基因的编辑效果。
目前常用的筛选方法包括PCR、限制性酶切和测序等。
验证编辑效果的方法则要根据具体实验目的来选择,比如功能研究可以采用功能分析、荧光染色或功能性检测等方法。
在实际应用CRISPR系统进行基因编辑时,还需注意以下几点:第一,选择目标基因时要明确实验目的,选择可行且有意义的目标。

CRISPRCas9基因敲除细胞株详细构建流程Puro 抗性浓度摸索实验将细胞如下图1稀释。
给药7天后,弃培养液,用台盼蓝染色2 min,显微镜下观察细胞存活情况。
确定细胞多克隆细胞筛选和单克隆细胞筛选浓度。
图1 抗性浓度摸索单克隆形成验证实验细胞计数,将细胞悬液稀释混匀后加入96孔板,用封口胶将孔板封好,放于培养箱中培养。
静置培养48 h后每日观察并记录单克隆形成情况。
sgRNA 靶标设计根据基因序列信息,设计 sgRNA。
靶标位点序列信息确认PCR扩增(图2),测序验证基因序列,以确定sgRNA区域有无SNP。
图2 靶标序列扩增sgRNA克隆引物合成根据sgRNA设计sgRNA克隆引物。
lentiCRISPRv2-sgRNA载体构建退火,连接,转化,涂板(LB/Amp)培养。
lentiCRISPRv2-sgRNA载体验证每个实验组各挑取6个单克隆菌落,于LB/ Amp培养基中扩增,提取质粒,琼脂糖凝胶电泳检测质粒抽提效果(图3)。
图3 质粒抽提酶切验证:取3个单克隆进行酶切,琼脂糖凝胶电泳检测酶切效果(图4)。
选择2个样品送样测序。
图4 单克隆酶切验证病毒包装lentiCRISPRv2-sgRNA无内毒素质粒提取,病毒包装。
细胞转染配制梯度病毒稀释液,细胞于培养箱中静置培养48h。
阳性单克隆细胞株筛选细胞转染48h后,更换完全培养基,筛选至对照组大部分细胞死亡,实验组细胞扩大培养,进行单克隆筛选。
几天后挑选阳性单克隆进行扩增,并取样验证。
阳性单克隆细胞株验证测序验证阳性单克隆细胞株的基因序列,以确定是否敲除成功。
实验结果示例:该基因有两个单克隆细胞株,A1和A2。
单克隆细胞A1的目的基因在sgRNA2位置出现两种突变形式,分别缺失1个和19个碱基,在新序列的第337位和第355位碱基提前出现终止密码子。
单克隆细胞A2的目的基因在sgRNA2位置发生突变,插入1个碱基,在新序列的第340位碱基提前出现终止密码子。

一、材料试剂准备1)质粒:pSpCas9(BB)-2A-GFP (Addgene plasmid ID: 48138),用于转染cas9,该质粒含有Cas9与GFP,Cas9的nickase活性将用于特定目的基因的ko,GFP可做为转染标签。
pSpCas9(BB) (Addgene plasmid ID: 42230),若实验用sgRNA为PCR扩增纯化产物,则需要该质粒做为U6的模版。
pUC19(Invitrogen, cat.no.15364-011)可用于构建sgRNA,若用PCR产物进行转染,则需要该质粒来进行共转染,做为DNA carrier。
上述三种质粒,根据实验选择sgRNA的表达方式(PCR产物/ 单载体系统/ 双载体系统)来确定具体使用哪种。
2)超纯水,DNase/RNase-free (Life Technologies, cat. no. 10977-023)3)高保真聚合酶,Kapa HiFi (Kapa Biosystems), PfuUltra (Agilen),Herculase II fusion polymerase 均可,只需保真效果好,扩增过程不产生突变。
4)Taq DNA polymerase with standard Taq buffer (NEB, cat. no. M0273S)用于一般检测。
5)QIAquick gel extraction kit (Qiagen, cat. no. 28704)6)QIAprep spin miniprep kit (Qiagen, cat. no. 27106)7)Fast Digest BbsI (BpiI) (Fermentas/Thermo Scientific, cat. no. FD1014),如需要将sgRNA构建到pSpCas9(BB)-2A-GFP质粒上,则需要该酶。
8)T7 DNA ligase with 2× rapid ligation buffer (Enzymatics, cat. no. L602L). 或者T4 DNA ligase,二者无区别。

crispr的实验流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!CRISPR 是一种基因编辑技术,以下是一般的实验流程:1. 设计 sgRNA:根据目标基因的序列,设计特异性的 sgRNA(向导RNA)。

CRISPR实验流程CRISPR/Cas9 实验流程交流企鹅: 2621697472(大家做哪块?)一、利用Cas9质粒建立knock-out细胞系实验的详细过程1.1 确定待敲除基因的靶位点1.2 设计识别靶位点的识别的一对DNA Oligo(引物)1.3 构建表达sgRNA的质粒1.4 sgRNA活性检测1.5 利用Cas9质粒建立knock-out细胞系1.1、确定待敲除基因的靶位点根据提供的物种、基因名称或者基因ID在NCBI或ENSEMBLE中进行查找。
找到该基因CDS区,分析相应的基因组结构,明确CDS的外显子部分。
按照基因本身的性质,选择候选的待敲除位点,确定待敲除位点。
对于蛋白编码基因,如果该蛋白具重要结构功能域,可考虑将基因敲除位点设计在编码该结构域的外显子;如果不能确定基因产物性质,可选择将待敲除位点放在起始密码子ATG后的外显子上。
如果是microRNA,可以将待敲除位点设计在编码成熟microRNA的外显子或在编码成熟microRNA的外显子的5’和3’侧翼序列。
1.2、设计识别靶位点的一对DNA Oligos确定待敲除位点后,选择23-至250bp的外显子序列输入到在线免费设计sgRNA的软件Input框中(/),然后进行设计运算,软件会自动输出sgRNA序列(网站设计一般很慢或数据输出不完整,可使用我的内部软件,2天内输出全部结果,无物种限制)。
一般地,基因特异的sgRNA模板序列为位于PAM序列(Protospacer Adjacent Motif)前间区序列邻近基序,这是一种见于crRNA分子的短核苷酸基序,可以被Cas9蛋白特异性识别并切割)的20个nt。
而PAM序列的特征为NGG(其中N为任意核苷酸)。
因此,sgRNA模板序列选择非常方便,即使没有软件,研究者也可手工进行选择。
不过,在线软件可以给出该序列在基因组中存在相似序列的情况,即可能的脱靶位点。
因此,利用在线软件可以选择脱靶机会小的序列作为sgRNA模板序列。
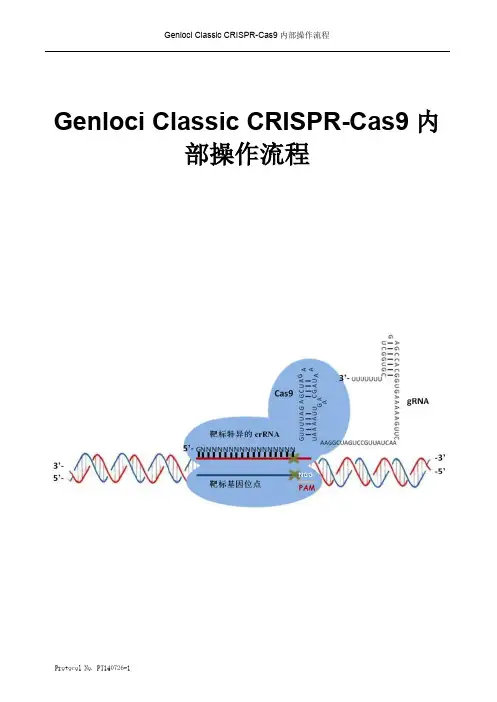
Genloci Classic CRISPR-Cas9内部操作流程Protocol No. PT140726-1Protocol No. PT140726-11,CRISPR-Cas9基因编辑实验流程图如下:pGK1.1U6 CACC G CAAA 4 55’ -5’3’-3’ -- ~20bp2,操作步骤实验前,请您务必做好以下验证实验:A.单细胞生长情况,确保单个细胞可以正常生长形成单克隆,即,低密度的细胞在培养皿中可以形成单克隆。
B.目的基因的表达情况分析,为防止因染色体缺失等情况导致靶基因缺失,首先需要PCR扩增靶基因,并对PCR结果进行测序,确保靶基因的完整存在;其次,您还可以用RT-PCR分析靶基因的活跃度。
1. 设计Oligo DNA序列首先,您需要在靶标DNA区域中设计一对20bp左右的,您可以通过以下在线工具设计:●麻省理工学院的CRISPR Design:/●德国癌症研究中心的E-Crisp:/E-CRISP/designcrispr下面我们选择麻省理工学院的CRISPR Design工具来做设计举例,以Fut8基因为例,一次只能输入大小为23~250bp的基因片段,最好一次只输入一个外显子,避免Guide序列跨内含子的。
点击“Download as genbank”按钮,出现以下界面:“Fut8”根据左边的score的高低选取合适的Guide序列,以Guide#1序列为例,2条单链oligo的序列如下(红色字体部分是要与Bbs I酶切后的载体相互补的部分):Fut8-F: cacc G AATGAGCATAATCCAACGCCFut8-R: aaac GGCGTTGGATTATGCTCATTC※注意:oligo DNA设计序列的第一个碱基必须是G,如果你选取的Guide序列的第一个碱基不是G,可自行加一个G上去。
另外,需在位点上下游各设计一条引物,用于后续PCR或测序检测阳性克隆,引物能扩增约300bp 的DNA片段,上游引物距突变位点约100bp,下游引物距突变位点约200bp。

CRISPR/Cas9敲除细胞系构建步骤及方法一、技术简介CRISPR/Cas9是最新出现的一种由RNA指导的Cas9核酸酶对靶向基因进行编辑的技术,也是目前研究最热的基因编辑技术。
由于其具有构建方法简单快捷、基因修饰效率高、成本低廉、实验周期短、适用范围广等诸多优点,目前已成功应用于人类细胞、斑马鱼、大/小鼠等多种动植物的基因组精确修饰。
二、实验流程1. 预实验1.1 Cas9导入细胞方法:尝试各种方法,如脂质体类转染、电转、慢病毒感染、腺病毒感染等,确定高效导入Cas9方法。
1.2 药物浓度预实验:降低后续阳性克隆筛选和检测工作难度。
1.3 单克隆培养情况:确认细胞是否可以单克隆培养。
2. 基因敲除(敲入)2.1 靶点设计:一般在不同转录产物的共同外显子上设计3个靶点,靶点位置尽量在基因CDS的前1/3,ATG之后,最好能破坏重要的domain和所有的转录产物isoform。
第一批合成构建3个,效果不佳或时间紧张的可一次构建6个。
2.2 载体构建和病毒包装:根据预实验结果,选择合适的普通载体或病毒载体(普通Cas9载体、慢病毒Cas9载体和腺病毒Cas9载体)。
2.3 内源活性筛选:转染细胞或感染细胞48h后,使用Puro或Blasticidin筛选48h,提取基因组DNA。
使用T7E1酶验证打靶载体的活性,将有效的突变型PCR产物测序验证。
2.4 Donor载体(基因敲入):根据筛选的gRNA靶点位置,构建Donor普通载体或腺病毒载体,共转染/感染Cas9-gRNA和Donor。
2.5 单克隆筛选:无限稀释到每孔1个细胞的数量,每株细胞铺至少2个96孔板。
细胞数量足够后,验证内源活性并送测。
2.6 获得突变型:如需纯合子,则可能需要重复步骤3-5。

crispr基因编辑技术流程英文回答:CRISPR Gene Editing Process.CRISPR gene editing is a powerful technique that allows scientists to make precise changes to DNA. The process involves using a guide RNA molecule to direct a Cas9 protein to a specific target gene. Once the target gene is found, the Cas9 protein will cut the DNA, allowing scientists to insert or remove genetic material.The CRISPR gene editing process can be divided into the following steps:1. Design a guide RNA molecule that is complementary to the target gene.2. Deliver the guide RNA molecule and the Cas9 protein to the cells.3. Allow the Cas9 protein to bind to the target gene.4. Cut the DNA at the target site.5. Insert or remove genetic material.6. Verify that the gene has been edited correctly.CRISPR gene editing has a wide range of potential applications, including:Treating genetic diseases.Developing new therapies for cancer.Creating new agricultural products.Improving our understanding of basic biology.CRISPR Gene Editing: A Closer Look.CRISPR gene editing is a revolutionary technology that has the potential to change the world. By allowing scientists to make precise changes to DNA, CRISPR could lead to new cures for diseases, new ways to improve crops, and new insights into how life works.Here's a closer look at how CRISPR gene editing works:CRISPR stands for Clustered Regularly Interspaced Short Palindromic Repeats. These are DNA sequences that are found in bacteria and archaea. CRISPR sequences are used by these organisms to defend themselves against viruses.CRISPR-Cas9 is a system that uses CRISPR sequences to guide a Cas9 protein to a specific target gene. Cas9 is a nuclease, which means that it can cut DNA.To use CRISPR-Cas9 for gene editing, scientists first design a guide RNA molecule that is complementary to the target gene. The guide RNA molecule is then delivered to the cells, along with the Cas9 protein.The Cas9 protein binds to the guide RNA molecule and uses it to find the target gene. Once the target gene is found, the Cas9 protein cuts the DNA at the target site.Scientists can then insert or remove genetic material at the target site. This can be used to correct genetic defects, introduce new genes, or disrupt genes that are involved in disease.CRISPR gene editing is a powerful tool that has the potential to revolutionize medicine, agriculture, and other fields. However, it is important to remember that CRISPR is still a new technology, and there are still some safety concerns that need to be addressed.CRISPR Gene Editing: Endless Possibilities.CRISPR gene editing has opened up a whole new world of possibilities for scientists to explore. With its ability to make precise changes to DNA, CRISPR could lead to new treatments for diseases, new ways to improve crops, and new insights into how life works.Here are just a few of the potential applications of CRISPR gene editing:Treating genetic diseases: CRISPR could be used to correct genetic defects that cause diseases such as sickle cell anemia, cystic fibrosis, and Huntington's disease.Developing new therapies for cancer: CRISPR could be used to develop new therapies for cancer that are more effective and less toxic than current treatments.Creating new agricultural products: CRISPR could be used to create new agricultural products that are more nutritious, resistant to pests and diseases, and better adapted to climate change.Improving our understanding of basic biology: CRISPR could be used to study how genes work and how they interact with each other. This could lead to new insights into how life works and how to treat diseases.The potential applications of CRISPR gene editing are endless. As scientists continue to explore this new technology, we can expect to see even more amazing things in the years to come.中文回答:CRISPR基因编辑技术流程。
Genloci Classic CRISPR-Cas9内部操作流程Protocol No. PT140726-1Protocol No. PT140726-11,CRISPR-Cas9基因编辑实验流程图如下:pGK1.1U6 CACC G CAAA 4 55’ -5’3’-3’ -- ~20bp2,操作步骤实验前,请您务必做好以下验证实验:A.单细胞生长情况,确保单个细胞可以正常生长形成单克隆,即,低密度的细胞在培养皿中可以形成单克隆。
B.目的基因的表达情况分析,为防止因染色体缺失等情况导致靶基因缺失,首先需要PCR扩增靶基因,并对PCR结果进行测序,确保靶基因的完整存在;其次,您还可以用RT-PCR分析靶基因的活跃度。
1. 设计Oligo DNA序列首先,您需要在靶标DNA区域中设计一对20bp左右的oligo DNA,您可以通过以下在线工具设计:●麻省理工学院的CRISPR Design:/●德国癌症研究中心的E-Crisp:/E-CRISP/designcrispr下面我们选择麻省理工学院的CRISPR Design工具来做设计举例,以Fut8基因为例,一次只能输入大小为23~250bp的基因片段,最好一次只输入一个外显子,避免Guide序列跨内含子的。
点击“Download as genbank”按钮,出现以下界面:“Fut8”Array根据左边的score的高低选取合适的Guide序列,以Guide#1序列为例,2条单链oligo的序列如下(红色字体部分是要与Bbs I酶切后的载体相互补的部分):Fut8-F: cacc G AATGAGCATAATCCAACGCCFut8-R: aaac GGCGTTGGATTATGCTCATTC※注意:oligo DNA设计序列的第一个碱基必须是G,如果你选取的Guide序列的第一个碱基不是G,可自行加一个G上去。
另外,需在位点上下游各设计一条引物,用于后续PCR或测序检测阳性克隆,引物能扩增约300bp 的DNA片段,上游引物距突变位点约100bp,下游引物距突变位点约200bp。
CRISPRCas9基因敲入试验步骤(三)donor载体构建载体构建1、质粒骨架的选择CRISPR/Cas9质粒按编辑细胞类型可分为八种,Mammalian 、Bacteria 、Drosophila 、Plant、C. elegans 、Yeast 、Zebrafish 、Xenopus,根据质粒所携带的编辑基因的不同,可分为野生型的cas9、突变型的cas9、cpf1、C2c2(可剪切RNA)。
当然,还有一些筛选标记,如puro、GFP、RFP、mcherry、潮霉素等等,我们可以根据自己的需求选择自己合适的质粒。
这里故意还掉了一种,最具有争议的Ngago,本楼主也重复过这个试验,也和大多数重复的人一样,GFP验证试验时,看到荧光显著减弱(本人还特意构建了一种Ngago载体,效果比韩老师的效果好很多),但是当时没有验证这个减弱是切割还是敲低,非常遗憾自己想法太简单,以先入为主认为是切割而没有去验证;最终结果是刘东老师使用Ngago发现了有表型(斑马鱼的眼睛上有缺陷),这个就是很强的提示,需要去做下一步,尽管基因组上没有被切割,有表型,那势必会去看该基因表达的数据,最后才有这篇文章。
刘东老师运气也很好,knock down 有表型,如果没表型,估计也会放弃。
这里就说到这,有不懂的可以留言询问。
具体分类在addgene上有,网址/。
2、酶切位点的选择插入sgRNA的酶切位点,质粒基本为这两种,BsmbI和Bbsi,具体可以参见该质粒的protocol,后续的连接、转化、验证protocol里面有,我就不啰嗦了。
哦还忘了一件事,使用snapgene这个软件可以很好地看质粒图谱,非常方便,强烈推荐!!以慢病毒载体V2为例具体说明3、同源臂的设计同源臂我们一般都是在切割位点的两端各选一段,长度大约1kb-2kb,效率还可以。
当然了,有人也用40-100bp做了也做成功了,但基本原则是越长效率越高。
1kb-2kb效率已经很高,所以这个最合适。
基因敲除的基本流程1. 设计基因敲除实验基因敲除实验首先需要设计实验,确定要敲除的基因以及敲除方式。
敲除方式包括siRNA敲除和CRISPR/Cas9敲除。
siRNA敲除是通过向细胞内注射siRNA分子来使目标基因表达受到抑制,从而实现基因敲除的目的。
而CRISPR/Cas9敲除则是通过设计引物,将Cas9质体和sgRNA质体共转染到目标细胞中,使得Cas9能够与sgRNA的引物匹配,精确地切割目标基因,从而实现基因敲除的目的。
2. 设计敲除载体敲除载体是进行基因敲除实验所必需的。
它是一种携带着基因敲除需要的siRNA或CRISPR/Cas9等转录因子,并且能够与目标细胞特定的受体结合,从而实现转录因子的进入细胞的载体。
根据实验的需要,可以选择合适的载体类型,包括质粒、病毒、脂质体等。
3. 转染转染是将敲除载体导入目标细胞的过程。
转染通常采用细胞培养技术,将载体与目标细胞放入培养皿中,在培养皿内进行培养。
转染通常需要一些转染试剂,以帮助载体更好地进入目标细胞。
4. 检测基因敲除效果一旦敲除载体进入了目标细胞,就需要进行基因敲除效果的检测。
这可以通过不同的方法来实现,例如PCR,PCR-引物肌积,蛋白质印迹等。
通过这些检测方法,可以明确敲除效果是否成功,如果成功,继续进入下一步骤。
5. 细胞筛选在基因敲除实验中,不是所有的细胞都会成功地被敲除,有些会发生杂交。
因此,需要通过细胞筛选的方法,将被敲除的细胞从未被敲除的细胞中分离出来。
这可以通过选择特定表型细胞、筛选无菌培养细胞、或选择对敲除载体特异性抗生素的细胞等方法来实现。
6. 确认敲除的稳定性确认基因敲除的稳定性,有多种方法,比如西方印迹法,北方印迹法等。
这些方法可以确定敲除的效果是否一直存在于筛选的细胞中,在不同的检测时间点,检测敲除效果是否保持可靠和稳定。
通过以上的基因敲除流程,就可以得出基因敲除的结论和结果。
可以为相关领域的研究和开发提供有价值的基础数据。
CRISPR实验流程
1.设计gRNA序列:
在CRISPR实验中,首先需要设计一个特定的gRNA(guide RNA)序列,用于引导Cas9酶找到并切割目标DNA序列。
gRNA通常由20个核苷酸组成,其中包含一个固定序列和一个可变序列。
可变序列需要与目标DNA序列中的特定区域互补配对。
2.合成gRNA:
设计好gRNA序列后,实验者可以选择将其合成或通过基因克隆技术将其插入表达载体中。
合成或构建gRNA的方法可以根据实验需求和实验室条件来选择。
3. 构建gRNA-Cas9表达载体:
将合成或构建好的gRNA序列插入Cas9表达载体中,以便在细胞中同时表达Cas9酶和gRNA。
这一步通常利用限制性内切酶或其他DNA重组技术进行。
4.细胞培养和转染:
5.目标DNA的识别和切割:
Cas9酶会与gRNA形成复合物,然后通过识别目标DNA序列的可变序列,与其互补配对。
一旦Cas9-gRNA复合物与目标DNA配对成功,Cas9酶将切割目标DNA,形成DNA双链断裂。
6.DNA修复:
DNA双链断裂会引发细胞的自修复机制,通常有两种DNA修复途径:非同源末端连接(NHEJ)和精准替代(HDR)。
NHEJ是一个错误拼接的过程,导致目标DNA序列的插入或缺失,从而引发基因功能发生改变。
而HDR则是通过提供外源DNA模板,并利用该模板进行精准修复的过程,可以实现比NHEJ更精确的DNA修复。
crispr_cas9细胞株构建流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor.I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!CRISPR/Cas9基因编辑技术:细胞株构建的详细流程CRISPR/Cas9系统,作为一种革命性的基因编辑工具,已经在生命科学研究和临床应用中发挥了巨大作用。
CRISPR/Cas9 实验流程
交流企鹅: 2621697472(大家做哪块?)
一、利用Cas9质粒建立knock-out细胞系实验的详细过程
1.1 确定待敲除基因的靶位点
1.2 设计识别靶位点的识别的一对DNA Oligo(引物)
1.3 构建表达sgRNA的质粒
1.4 sgRNA活性检测
1.5 利用Cas9质粒建立knock-out细胞系
1.1、确定待敲除基因的靶位点
根据提供的物种、基因名称或者基因ID在NCBI或ENSEMBLE中进行查找。
找到该基因CDS区,分析相应的基因组结构,明确CDS的外显子部分。
按照基因本身的性质,选择候选的待敲除位点,确定待敲除位点。
对于蛋白编码基因,如果该蛋白具重要结构功能域,可考虑将基因敲除位点设计在编码该结构域的外显子;如果不能确定基因产物性质,可选择将待敲除位点放在起始密码子ATG后的外显子上。
如果是microRNA,可以将待敲除位点设计在编码成熟microRNA的外显子或在编码成熟microRNA的外显子的5’和3’侧翼序列。
1.2、设计识别靶位点的一对DNA Oligos
确定待敲除位点后,选择23-至250bp的外显子序列输入到在线免费设计sgRNA的软件Input框中(/),然后进行设计运算,软件会自动输出sgRNA序列(网站设计一般很慢或数据输出不完整,可使用我的内部软件,2天内输出全部结果,无物种限制)。
一般地,
基因特异的sgRNA模板序列为位于PAM序列(Protospacer Adjacent Motif)前间区序列邻近基序,这是一种见于crRNA分子的短核苷酸基序,可以被Cas9蛋白特异性识别并切割)的20个nt。
而PAM序列的特征为NGG(其中N为任意核苷酸)。
因此,sgRNA模板序列选择非常方便,即使没有软件,研究者也可手工进行选择。
不过,在线软件可以给出该序列在基因组中存在相似序列的情况,即可能的脱靶位点。
因此,利用在线软件可以选择脱靶机会小的序列作为sgRNA模板序列。
根据选择的sgRNA模板序列,合成一对序列互补的DNA Oligos(同时设计检测目的基因的引物一起合成)。
1.3、构建可表达sgRNA的质粒
(Precut SgRNA Cloning kit and pSD-gRNA Plasmid构建试剂盒)
企鹅: 2621697472
将合成的Oligos以逐步降温的方法退火成双链,然后与我们提供的质粒进行连接,连接后转化感受态的大肠杆菌,再进行涂板。
次日在LB琼脂平板上,挑取单克隆6个,溶于20ul LB液中,涡旋后,将部分菌液接种到LB液中,37oC下250 rpm生长,另部分的菌液用作模板进行快速PCR,经电泳确定阳性克隆后,将相对应的细菌送商业公司测序,同时将部分菌液接种到LB液体,37oC下250 rpm培养过夜。
1.4、sgRNA活性检测
1.4.1 sgRNA活性预检测--SSA活性检测
(Precut pSG-target Cloning kit & SSA assay检测试剂盒)
企鹅: 2621697472
• SSA检测:根据客户要求检测sgRNA的活性及敲除效率,客户也可
以选购我公司Precut pSG-target Cloning kit自行构建报告载体用于检测,我公司提供阳性及阴性sgRNA及其SSA report target质粒。
• 检测原理:SSA报告质粒(pSG-target)中luciferase基因被终止密码子提前终止,这种截短的luciferase没有活性。
为检测sgRNA的剪切活性,将Cas9/sgRNA的靶点序列插入到终止密码子之后。
在Cas9和sgRNA 的作用下,靶点位置的序列被有效剪切形成DSB,细胞通过同源重组重新形成有活性的luciferase(Figure 2),通过检测luciferase的活性升高就可以反应Cas9/sgRNA的剪切活性(Figure 3)。
Figure 2. Restore disrupted luciferase function via sgRNA-mediated homologous recombination
Figure 3.Increased FL/RL ratio in Cas9/sgRNA transfection cells.
1.4.2、CRISPR剪切活性检测--surveyor
CRISPR/Cas9打靶基因后引入的突变的方式与ZFN、TALEN基本一致,都是NHEJ或是HR。
因此在CRISPR/Cas9的应用中,活性检测或是突变效率的检测可以参照ZFN和TALEN方法.主要包括有:靶位点直接PCR后TA克隆测序和基于可以识别错配双链的错配内切酶检测法(Surveyor法)。
Surveyor法即错配酶法:靶序列经CAS/sgRNA切割后由于缺乏修复模板,将主要以非同源重组的方式进行修复,或多或少会插入或删除一些碱基。
因此将靶序列PCR扩增后经变性、退火,将形成错配。
错配酶(主要是CEL1或T7E1酶)将识别错配的杂合双链并剪切。
产物跑电泳,比较切割条带与未切割条带的比例,即可反映出Cas/sgRNA 的活性。
1.4、利用Cas9质粒建立knock-out细胞系
将Cas9质粒转染细胞,如果细胞用脂质体转染困难,可采用电转。
通过荧光标记观察转染效率,如果选择了带Neo抗性的质粒,则同时用G418药物筛选。
如果有FACS,可直接分选带荧光的细胞。
或者当细胞长满培养皿时,将细胞消化成单细胞,采用有限稀释法,接种96孔板。
根据前述突变率决定克隆接种数量,由于Indel突变是随机突变。
因此,其中的2/3应该是移码突变。
如突变率=30%,则建议接种克隆数为96个,最终约有5个阳性克隆。
在荧光显微镜下观测细胞克隆生长情况,选择带有荧光的克隆,适时
进行胰酶消化后,提取部分细胞的基因组DNA用作PCR模板。
然后以前述引物和PCR条件进行PCR扩增。
PCR产物先进行2%琼脂糖凝胶电泳,如果出现条带,则将PCR产物直接送测序。
测序时,PCR产物无需切胶。
待测序结果返回后,人工阅读测序色谱图。
可确定基因是否突变以及突变基因的基因型。
将携带有双突变等位基因的阳性克隆扩增,保存,稳转系建立成功。
二、利用Cas9质粒建立可遗传的基因敲除动物品系的详细实验过程:
2.1 确定待敲除基因的靶位点
2.2 设计识别靶位点的识别的一对DNA Oligo(引物)
2.3 构建可表达sgRNA的Cas9质粒
2.4 体外转录sgRNA 和Cas9 RNA
2.5 将sgRNA和Cas9 RNA直接注射入受精卵检测sgRNA活性
2.6 将有活性的sgRNA和Cas9 RNA直接注射入受精卵建立Founder 2.7 将Founder自交得到F1
2.8 F1自交得到F2
2.1 确定待敲除基因的靶位点
同前
2.2 设计识别靶位点的识别的一对DNA Oligo(引物)
同前
2.3 构建可表达sgRNA的Cas9质粒
同前,选择带有T7启动子的质粒
2.4 体外转录sgRNA 和Cas9 RNA
使用特定引物,以上述质粒为模板,以高保值酶分别对Cas9和sgRNA 进行PCR扩增,循环数设为30个,20 ul反应体系。
然后将产物纯化用作体外转录模板。
体外转录时,Cas9 RNA需要戴帽和加尾,而sgRNA 无需戴帽和加尾。
2.5 将sgRNA和Cas9 RNA直接注射入受精卵检测sgRNA活性
将体外转录的sgRNA和Cas9 RNA直接注射入斑马鱼受精卵中进行活性检测;对于非斑马鱼基因,将含待敲除靶位点序列的DNA片段进行扩增,然后和上述两种RNA共同注射入斑马鱼受精卵。
待斑马鱼胚胎发育至24小时,随机选取5个胚胎,提取基因组DNA,然后以此为模板进行PCR扩增。
然后按前面同样的方法进行sgRNA的活性验证。
2.6 将有活性的sgRNA和Cas9 RNA直接注射入受精卵建立Founder
将体外转录的有活性的sgRNA和Cas9 RNA直接注射入动物受精卵,获得基因敲除的初建者。
待初建者发育至可进行基因型鉴定时(斑马鱼为4个星期),剪取部分尾鳍,提取基因组DNA,作为PCR模板,然后按前面同样的方法进行基因突变鉴定。
选取有突变的斑马鱼用作传代实验。
4.7 将Founder自交得到F1
以斑马鱼为例,将阳性Founder自交,得到F1。
待F1生长至4星期时,进行剪取部分尾鳍,提取基因组DNA,作为PCR模板,然后按前面同样的方法进行基因突变鉴定。
选取有移码突变的斑马鱼用作建系。
8)F1自交得到F2
对于特定的基因,建立基因鉴定的酶切或PCR方法,通过将携带相同基因型突变等位基因的F1斑马鱼自交,获得纯合突变体、杂合突变体。
如果纯合突变不致死,则可将纯合突变F2传代,从而建立纯合突变体系。
如果纯合突变致死,则可将杂合突变F2传代,从而建立杂合突变体系。
动物实验推荐使用体外转录的sgRNA
(sgRNA体外转录试剂盒)
(一步纯化RNA转录试剂盒)
企鹅: 2621697472。