用于熔盐体系的氮化硼隔膜Ag_AgCl参比电极性能
- 格式:pdf
- 大小:526.97 KB
- 文档页数:5

镍掺杂石墨相氮化碳的熔盐辅助微波法制备及光催化固氮性能曲晓钰;胡绍争;李萍;王菲;赵艳锋;王琼【摘要】In this work, nickel doped g-C3N4 was synthesized via a novel molten salt-assisted microwave process. X-ray diffraction(XRD), N2 adsorption, UV-Vis spectroscopy, scanning electron microscopy(SEM), temperature-programmed desorption(TPD), X-ray electron spectroscopy(XPS), photoluminescence spectros-copy(PL) and electrochemical impedance spectroscopy(EIS) were used to characterize the prepared catalysts. The results show that the molten salt-assisted microwave process changes the morphology of prepared catalyst from layered structure to nanoparticles. These nanoparticles are closely packed with each other to form many secondary pores, which increases the catalyst surface area. Besides, due to that the raw materials are wrapped by the liquid-phase molten salt during the microwave process and can not be in contact with oxygen, Ni is not only present as inactive oxide but inserts at the interstitial position to form active Ni(Ⅰ)—N bonds. This Ni(Ⅰ)—N active sites can activate N2 molecules, promote separation rate of electrons and holes, and accelerate interfacial charge transfer from catalysts to N2 molecules, thus significantly improving the nitrogen photofixation ability.%采用熔盐辅助微波法制备了可见光下具有优越光催化固氮性能的镍掺杂石墨相氮化碳.采用X射线衍射(XRD)、扫描电镜(SEM)、氮气吸附-脱附、紫外-可见光谱(UV-Vis)、X射线光电子能谱(XPS)、荧光光谱(PL)、程序升温脱附(TPD)和电化学阻抗谱(EIS)等手段对催化剂进行了表征.结果表明,熔盐辅助微波法使氮化碳催化剂从层状结构变为纳米颗粒状,并相互紧密堆积形成很多二次孔,增大了催化剂的比表面积.同时,在催化剂制备过程中,熔盐包裹住了催化剂原料,避免了镍离子与氧气的接触,使镍离子呈现出活性的Ni(Ⅰ)—N态和非活性的氧化镍态2种存在形式.Ni(Ⅰ)—N作为反应活性中心,能有效捕获光电子,提高电子-空穴分离效率,促进电子从掺杂镍离子向N2分子的迅速转移,实现氮气分子的活化,进而提高固氮性能.【期刊名称】《高等学校化学学报》【年(卷),期】2017(038)012【总页数】9页(P2280-2288)【关键词】熔盐辅助微波合成;石墨相氮化碳;Ni—N活性位;光催化固氮【作者】曲晓钰;胡绍争;李萍;王菲;赵艳锋;王琼【作者单位】辽宁石油化工大学化学化工与环境学部, 抚顺 113001;辽宁石油化工大学化学化工与环境学部, 抚顺 113001;辽宁石油化工大学化学化工与环境学部, 抚顺 113001;辽宁石油化工大学化学化工与环境学部, 抚顺 113001;辽宁石油化工大学化学化工与环境学部, 抚顺 113001;辽宁石油化工大学化学化工与环境学部, 抚顺 113001【正文语种】中文【中图分类】O644氨作为富氢材料, 因具有能量密度高(氢质量分数为17.6%)和易于储运等优点而备受关注. 氮作为合成蛋白质的原料, 也是人类与许多其它生物生长发育所必需的元素. 虽然氮气占空气组成的78%, 但是其不能被绝大多数生物直接利用. 因此, 固氮作用是自然界中除光合作用外最重要的化学反应. 目前, 采用Haber法的工业固氮过程已经达到年产亿吨的水平. 此方法需要高温高压及氢气作为反应原料, 不仅反应条件苛刻, 原料价格昂贵, 还存在一定的危险性. 因此, 需要寻找一种绿色清洁、低能耗的固氮工艺来代替Haber法. 随着多相光催化技术的不断发展, 光催化固氮产氨技术受到了研究者广泛的关注[1~6]. 1977年, Schrauzcr等[1]首次发现在光照条件下氮气可以在Fe掺杂TiO2粉末上发生还原反应生成氨. 此后, Ranjit等[2]制备了贵金属负载的TiO2催化剂, 发现其光催化固氮性能与金属-氢键的强度有关, Ru负载的TiO2催化剂表现出最佳的固氮性能. Rusina等[3]制备了Fe2Ti2O7催化剂, 并实现了可见光下的光催化固氮反应. Hoshino等[4]制备了导电聚合物/TiO2复合催化剂. 光催化固氮反应结果显示, TiO2中氧空穴作为吸附、活化和还原氮气分子的反应中心, 其含量与固氮效果有直接关系. 然而由于光催化固氮是一个热力学上不能自发进行的反应, N≡N键能高达941 kJ/moL, 第一电离势为15.58 eV, 具有较高的稳定性, 不能像固氮酶一样实现在常温常压下将N2和H2O 转化成氨. 因此光催化固氮反应的关键在于N2分子的活化[5].石墨相氮化碳(g-C3N4)是一种有机非金属层状材料, 具有特殊的理化性质、优异的化学稳定性及可调变的电子结构[6~8]. 然而, 采用高温缩聚法制备的g-C3N4样品具有较大的晶粒尺寸, 导致比表面积很小, 只有约10 m2/g, 限制了g-C3N4在催化、光电材料等领域的应用[9]. 目前最常用的制备具有较大比表面积g-C3N4催化剂的方法是模板法, 即在合成体系中加入硬模板或有机模板剂, 后续再将此模板或模板剂焙烧或溶解除去. 此方法不仅污染环境, 耗材价格昂贵, 而且制备方法繁琐.微波加热不同于其它常规加热方式, 它通过被加热体内部偶极分子高频往复运动产生“内摩擦热”而使被加热物料温度升高, 不需要任何热传导过程就能使物料内外部达到同时升温的目的. Yu等[10]采用微波法在短时间内制备出高比表面积g-C3N4催化剂, 其表现出良好的催化性能. 该方法还具有能耗低、有害气体排放低及产物收率高等优点. 近年来, 熔盐法被广泛应用于材料合成领域. 此方法可以加速反应物离子的扩散, 控制产物晶体生长, 并且熔盐可通过水洗法很容易地从产物中分离出来[11~13]. 另外, 熔盐法能最小化固体产物与熔盐间的界面能, 从而形成特殊的产物形貌. 然而常规的电阻热熔盐法存在耗时长、能耗高、产物收率低的问题. 将微波法与熔盐法的优点相结合是制备高性能氮化碳催化剂的理想方法.研究发现, BiOBr、 g-C3N4和复合金属硫化物中的氧空穴、氮空穴和硫空穴可以作为反应活性中心, 一方面与氮气间产生强化学吸附, 活化N≡N键; 另一方面能捕获光电子, 并促进光电子从催化剂向氮气分子转移, 显著提高固氮性能[14~16]. 由此可设想, 掺杂金属阳离子作为另一种表面缺陷态, 也可能作为反应活性中心对固氮反应起类似的促进作用. 基于上述设想, 本文采用熔盐辅助微波法制备了具有较大比表面积的可见光下具有优越光催化固氮性能的镍离子掺杂石墨相氮化碳催化剂. 一方面, 镍离子掺杂可以有效捕获光电子, 提高分离效率, 并且基于过渡金属与N2间的电子反向赠与现象, 电子能实现从掺杂镍离子向N2分子的迅速转移[17~19]; 另一方面, 熔盐辅助微波法使氮化碳催化剂由层状结构变成小颗粒状, 并相互紧密堆积形成很多二次孔, 增大了催化剂的比表面积. 同时, 在催化剂制备过程中熔盐包裹住了催化剂原料, 有效避免了镍离子与氧气的接触, 使镍离子呈现出氧化镍和Ni(Ⅰ)—N 2种形态. 实验结果表明, Ni(Ⅰ)—N为催化剂的反应活性中心, 而氧化镍为非活性态.氯化镍、双氰胺、氯化锂、氯化钾、氧化铜均为分析纯, 购自天津化学试剂厂, 使用前未经提纯. 所有水溶液均用去离子水配制.XRD-7000 X射线衍射仪(XRD, 日本岛津公司, Cu Kα1靶, 工作电压40 kV, 工作电流30 mA); ASAP 2010物理吸附仪(美国Micrometrics公司, 测试温度-196 ℃, 测试前样品先于350 ℃下真空脱气处理10 h); JSM 5600LV扫描电子显微镜(SEM,日本电子有限公司); CHEMBET-3000程序升温脱附仪(TPD, 美国康塔仪器公司); Thermo ESCALAB 250光电子能谱仪(XPS, 美国赛默飞世尔科技有限公司);Elan6100DRC型电感耦合等离子体质谱仪(ICP-MS, 美国Perkin Elmer公司); UV-550紫外-可见光谱仪(UV-Vis, 日本JASCA公司); FP-6300荧光分光光度计(PL, 日本分光公司, 以Xe灯为激发光源); AUTO-LAB PGSTAT30电化学工作站(瑞士万通公司), 测试在自制带石英窗口的电解池(20 mm×40 mm×50 mm) 中进行, 采用三电极体系, 以膜状样品为工作电极, Pt片为对电极, Ag/AgCl为参比电极, 扫描频率范围为1~100 MHz, 控制在开路电位, 扰动电位为l0 mV, 电解质为0.5 mol/L Na2SO4.将6 g双氰胺与一定量的NiCl2·6H2O(镍/双氰胺摩尔比为0.005)加入到20 mL乙醇中. 于60 ℃加热蒸干乙醇后, 将得到的固体置于坩埚中, 加入质量比1∶1的KCl-LiCl混合物(KCl-LiCl/双氰胺质量比为50∶1), 将此坩埚放入200 mL大坩埚中, 并用微波吸收材料CuO粉末填满大坩埚. 将大坩埚放入微波炉中(G70D20CN1P-D2, 格兰仕公司), 微波处理30 min, 输入功率1.0 kW/h. 得到的催化剂命名为NiCN(MS). 重复上述方法, 不加入NiCl2·6H2O, 得到的催化剂命名为CN(MS); 不加入KCl-LiCl混合物, 得到的催化剂命名为NiCN(MV); 不加入NiCl2·6H2O及KCl-LiCl混合物, 得到的催化剂命名为CN(MV). 为研究Ni的存在形式对催化性能的影响, 将制备的NiCN(MS)在空气中于550 ℃分别焙烧1, 2, 3 h, 得到的催化剂命名为NiCN(MS)-1, NiCN(MS)-2及NiCN(MS)-3.根据文献[20]报道的方法评价催化剂的光催化固氮性能. 以250 W高压钠灯(主波长400~800 nm)为可见光光源, 以0.5 mol/L的亚硝酸钠水溶液为钠灯冷却循环水滤去光源中紫外光部分(流速0.5 L/min). 以0.789 g/L的乙醇水溶液为空穴捕获剂. 将0.2 g催化剂加入到500 mL乙醇水溶液中, 搅拌0.5 h以充分分散. 然后在搅拌条件下将溶液置于光源下进行照射, 同时鼓入空气(30 ℃, 标准大气压). 每隔1h取5 mL悬浮液于离心管中, 在转速为3000 r/min的条件下离心15 min后取上层清液, 利用紫外-可见分光光度计, 采用纳氏试剂法[5,20]分析产物中铵离子浓度. 具体方法如下: 取10 mL待测样品放入25 mL容量瓶中并定容到25 mL. 滴加0.5 mL酒石酸钾钠溶液(2.17 mol/L)和0.75 mL纳氏试剂, 静置10 min后, 采用紫外-可见分光光度计测试420 nm波长下的吸光度. 铵离子浓度(mg/L)采用标准曲线换算得到, 换算公式为: 铵离子浓度=(试样吸光度-空白样吸光度)×5.642.由图1可见, 在没有光照、氮气和催化剂的条件下, 铵离子产率均可忽略不计, 说明铵离子是以氮气为原料通过光催化还原制得的. 由图2可以看出, 反应1 h时,CN(MV)的铵离子产率非常低, 只有熔盐辅助微波法制备的CN(MS)铵离子产率略有提高, 为引入Ni后, NiCN(MV)的铵离子产率仅为而NiCN(MS)的铵离子产率高达分别是CN(MV), CN(MS)和NiCN(MV)的20.9倍、 15.6倍和9.4倍. 可见, 熔盐辅助微波法可使NiCN(MS)中的Ni产生不同于NiCN(MV)中Ni的存在形式, 导致两者固氮性能产生巨大差异. 此外, 图2插图显示, NiCN(MS)催化剂经过40 h的长时间反应后, 产氨效率几乎不变, 说明此催化剂具有优异的催化稳定性. 通过ICP-MS测得NiCN(MS)的镍离子含量在反应前后没有显著变化(质量分数分别为0.20%和0.19%), 说明催化剂在结构上也很稳定. 采用熔盐辅助微波法制备Ni掺杂氮化碳催化剂过程中, 镍与双氰胺摩尔比为0.002, 0.005, 0.010和0.020时, 所得催化剂的铵离子产率分别为2.9, 6.7, 5.8和镍与双氰胺的摩尔比增加至0.005时, 固氮性能达到最佳, 继续提高镍与双氰胺的摩尔比后, 固氮性能反而降低. 由此得到最佳镍/双氰胺摩尔比为0.005.图3示出了反应体系pH值对CN(MS)光催化固氮的影响. 可以看出, 随着反应体系pH值的提高, 催化剂的固氮能力逐渐下降. 这是由于酸性条件下, 体系中的H+能促进氮气分子的转化, 而碱性条件提供的OH-却不利于固氮反应进行. 在反应体系中加入电子捕获剂AgNO3后, NiCN(MS)的固氮能力显著下降[图4(A)], 而用疏质子溶剂DMF和DMSO代替水进行反应时则几乎没有铵离子生成[图4(B)]. 这说明电子是反应活性物种, 而水为还原反应提供了质子, 反应式如下:图5示出了反应体系的pH值随反应时间的变化情况. 反应开始时体系pH值为6.2, 呈弱酸性. 经过24 h反应后体系的pH值升高到了8.0, 这是由于固氮反应消耗了水中的氢离子所致. 此外, 按照文献[4]方法制备了氮空穴掺杂的g-C3N4催化剂(V-g-C3N4), 并比较了V-g-C3N4与NiCN(MS)的固氮性能. 由图6可见, NiCN(MS)的固氮性能与V-g-C3N4相当. 40 h催化稳定性结果[图6(B)]显示, NiCN(MS)的固氮性能没有显著变化, 而V-g-C3N4的铵离子产率显著降低, 这可能是由于氮空穴作为反应活性位其结构不稳定所致.图7为所制备催化剂的SEM照片. 可以看出, 微波法制备的CN(MV)为无规则形貌的片状结构[图7(A)]. Ni引入后得到的NiCN(MV)的形貌与CN(MV)相比没有显著变化[图7(B)]. 而熔盐辅助微波法制备的CN(MS)和NiCN(MS)的形貌均为由纳米颗粒密集堆积而成的块状结构[图7(C), (D)]. 由此可知, 熔盐的引入对催化剂的形貌有显著影响, 这与文献[21~23]的研究结果一致.图8(A)为所制备催化剂的XRD谱图. 图中有2个特征峰, 分别位于2θ=13.1°和27.5°处, 其中2θ=27.5°处的衍射峰为芳香物层间堆积特征峰, 对应于(002)晶面[24]; 2θ=13.1°处的衍射峰是Melon类结构的特征峰, 对应于(100)晶面. 图中没有发现与镍物质有关的衍射锋, 说明镍均匀分散在氮化碳催化剂中. 此外, 熔盐辅助法制备的2个催化剂的衍射锋强度与单纯微波法得到的催化剂相比显著降低, 说明熔盐辅助法制备的氮化碳具有更小的粒径尺寸, 这与SEM的结果相一致. 图8(B)为所制备催化剂的UV-Vis光谱. 可见, CN(MV)和CN(MS)显示了石墨相氮化碳类半导体的典型吸收光谱, 其吸收边界为460 nm. 引入镍后, NiCN(MV)和NiCN(MS)的吸收边界略有位移, 对可见光的吸收也有所增强. 这是由于镍离子掺杂后改变了催化剂的电子结构所致[25]. 根据 Kubelka-Munk方程计算了样品的带隙能[图8(C),α为吸光度, h为普朗克常数, ν为光频率], CN(MV)和CN(MS)的带隙能为2.70 eV, 而镍掺杂后NiCN(MV)和NiCN(MS)的带隙能减小到2.61 eV. 图8(D)为所制备催化剂的N2吸附-脱附等温线. 如图所示, 制备的催化剂均为Ⅳ型等温线. 计算结果表明, CN(MV)和NiCN(MV)的比表面积很小, 分别只有8.8和9.2 m2/g; 熔盐辅助法制备的CN(MS)和NiCN(MS)的比表面积显著提高, 分别达到22.4和24.5 m2/g. 这是由于熔盐辅助法制备的氮化碳纳米颗粒相互堆积, 形成了二次孔导致的. 催化剂比表面积的提高能使更多的反应活性中心暴露出来, 利于更多的反应物同时进行反应, 对催化剂性能有重要的影响.XPS是表征催化剂表面元素状态的有效手段. 图9(A)中CN(MV)和NiCN(MS)的C1s轨道有2个峰, 其中284.6 eV处的峰归属为环状结构中sp2杂化的C原子(N―CN), 287.9 eV处的峰则归属为石墨型结构中C—N键中的C原子[26,27]. 图9(B)为所制备催化剂的N1s轨道XPS谱图. CN(MV)和NiCN(MS)的2个结合能位于398.4和400.2 eV附近, 分别归属于sp2杂化的N原子(C―NC)和芳香环中连接3个C原子的N原子[28]. 如图9(C)所示, NiCN(MV)中Ni的结合能位于857.5 eV处, 应归属于NiO[29,30]. NiCN(MS)的Ni2p轨道出现2个结合能, 除了857.5 eV处的NiO以外, 851.9 eV处的结合能根据文献[31]报道可能归属于+1价的Ni+与氮结合得到的Ni(Ⅰ)—N键. 因此推断Ni在NiCN(MS)中一部分以氧化态存在; 另一部分插入了氮化碳的晶格间隙, 由于氮化碳为富电子结构, Ni+与N的孤对电子形成Ni(Ⅰ)—N配位键. 这可能是由于催化剂制备过程中熔盐包裹住了催化剂原料, 尽量避免了镍离子与氧气的接触, 使镍离子呈现出氧化镍和Ni(Ⅰ)—N 2种形态. 此外, Ma等[32]的理论计算结果也表明, 氮化碳晶格中氮孔的最大平面间距为0.71 nm, 远大于Ni+的半径(0.07 nm), 说明Ni+晶格间隙掺杂在理论上也是可行的. 结合图2中NiCN(MV)和NiCN(MS)固氮性能的差异可以推测, NiCN(MS)催化剂中的反应活性中心可能是Ni(Ⅰ)—N形态的Ni+, 而非NiO.图9(D)为CN(MV)和NiCN(MS)的VB XPS结果. CN(MV)的EVB位于1.40 eV 处; NiCN(MS)的EVB位于1.52 eV处, 与CN(MV)相比发生了明显的位移, 这可能是由于Ni+掺杂引起氮化碳电子排布变化所导致的. 结合UV-Vis的结果可以推断CN(MV)和NiCN(MS)的ECB分别位于-1.3 eV和-1.09 eV处. 此结果说明CN(MV)上的光电子应该比NiCN(MS)的光电子具有更强的还原能力. 然而CN(MV)的光催化固氮能力却远不如NiCN(MS)催化剂. 由此可知Ni掺杂后催化剂光催化固氮性能的提高不是源自于Ni掺杂后催化剂能带位置的改变.在多相催化反应中, 化学吸附能活化反应物分子, 因此化学吸附位通常被认为是反应的活性中心. 利用N2-TPD表征了N2分子在催化剂表面的吸附情况, 结果如图10(A)所示. 由图中可看出, CN(MV)和NiCN(MV)表面只存在1种吸附的氮气物种, 其脱附温度约为110 ℃, 应为N2分子的物理吸附. 而NiCN(MS)催化剂表面存在2种吸附的氮气物种, 脱附温度分别为110和230 ℃. 由此可知, NiCN(MS)除了对氮气存在物理吸附作用外, 还存在化学吸附作用. NiCN(MS)在110 ℃的TPD峰面积远大于CN(MV)和NiCN(MV), 说明NiCN(MS)能吸附更多的N2分子, 应具有更大的比表面积, 这与BET的计算结果相一致. 此外, 焙烧时间越长, 230 ℃处的脱附峰越弱, NiCN(MS)对氮气分子的化学吸附能力越差. 结合XPS表征结果判断, 对N2分子起化学吸附作用的应为NiCN(MS)中的Ni(Ⅰ)—N活性位, 而非+2价的NiO. 为进一步验证此结果, 以XPS结果中焙烧前后的NiCN(MS)催化剂中 +1价Ni的峰面积与总Ni的峰面积比值为横坐标, 铵离子产率为纵坐标作图, 结果如图10(B)所示. 可以看出, 两者之间呈良好的线性关系, 相关度R=0.992. 此结果也说明了催化剂中对氮气分子起吸附和活化作用的为+1价态的Ni(Ⅰ)—N, 而非氧化态的Ni2+.光生电子-空穴对的有效分离能保证提供充足的光电子用于还原氮气分子. 图11(A)为所制备催化剂的电化学阻抗谱图. 图11中CN(MV)的圆弧半径最大, 表明其电荷转移电阻大, 不利于电荷传输. CN(MS)的阻抗弧半径略有减小, 可能是由于催化剂粒径尺寸减小缩短了光生电子-空穴从体相到表面的迁移距离, 降低了复合几率. NiCN(MS)的阻抗弧半径最小, 表示该催化剂具有最高效的界面电荷迁移速率, 能最有效地分离光生电子-空穴对. 此外, NiCN(MV)的阻抗弧半径远大于NiCN(MS), 说明促进光生电子-空穴对有效分离的是+1价态的Ni(Ⅰ)—N, 而非氧化态的Ni2+. 图11(B)为所制备催化剂在氮气气氛下的PL图谱. 所有催化剂均在460~470 nm 附近具有荧光发射峰, 与UV-Vis谱图中的吸收边界相一致. 与CN(MV)相比,CN(MS)的荧光发射峰强度略有降低, 而NiCN(MS)催化剂则发生了明显的荧光猝灭现象. 此外, NiCN(MV)的荧光发射峰强度远大于NiCN(MS),表明Ni(Ⅰ)—N活性位能捕获光电子, 促进光生电子-空穴对有效分离, 而氧化态的Ni2+为非活性态, 对催化性能无显著影响. 图11(C)比较了NiCN(MS)在氮气气氛下焙烧前后的PL光谱强度. 可以看出, 随着焙烧时间的延长, PL光谱强度逐渐增加. 这是由于焙烧过程使+1价态的Ni(Ⅰ)—N转变成氧化态的Ni2+, 失去了对电荷分离的促进作用. 图11(D)比较了CN(MV)与NiCN(MS)在氮气和氩气气氛下的PL光谱强度. 可见, CN(MV)在2种气氛下PL光谱强度基本一致, 而NiCN(MS)在氮气气氛下的PL强度明显低于氩气气氛下的光谱强度, 这说明氮气气氛对光生电子-空穴的分离起到了促进作用. 光电子可能从催化剂转移至氮气分子表面, 抑制了复合, 而Ar气气氛下则没有此电子转移现象, 因此对电子-空穴对的分离没有促进作用.推断可能的N2活化及光催化固氮反应机理如Scheme 1所示. 首先, 氮气化学吸附在Ni(Ⅰ)—N活性位上, 当催化剂被可见光激发后产生电子-空穴对(Step 1). 光电子被Ni(Ⅰ)—N活性位捕获, 并迅速转移至吸附的氮气表面(Step 2). 由于氮气分子的成键轨道已经被4个电子占满, 这个光电子只能占据高能态的反键轨道, 使氮气分子活化(Step 3). 活化的氮气分子与水中的H+反应生成NH3, 并溶于水中得到离子(Step 4). 在此过程中, Ni(Ⅰ)—N活性位一方面作为化学吸附位, 起到吸附并活化氮气分子的作用; 另一方面Ni(Ⅰ)—N活性位促进了光生电子从催化剂向氮气分子的转移, 抑制了电子-空穴对的复合, 因此对催化剂的光催化固氮性能有显著的促进作用.采用熔盐辅助微波法制备了具有较大比表面积并在可见光下具有较高光催化固氮性能的镍离子掺杂石墨相氮化碳催化剂. 催化剂制备过程中熔盐包裹住了催化剂原料, 避免了镍离子与氧气的接触, 使镍离子呈现出氧化镍和Ni(Ⅰ)—N 2种形态. 实验及表征结果表明, Ni(Ⅰ)—N为催化剂的反应活性中心, 而氧化镍为非活性态.Ni(Ⅰ)—N活性位能有效地捕获光电子, 提高电子-空穴分离效率, 促进电子从掺杂镍离子向N2分子的迅速转移. 此外, 熔盐辅助微波法改变了氮化碳催化剂形貌, 从层状结构变为纳米颗粒状, 并相互紧密堆积形成很多二次孔, 增大了催化剂的比表面积. NiCN(MS)的铵离子产率高达分别是CN(MV), CN(MS)和NiCN(MV)的20.9倍、 15.6倍和9.4倍, 且具有优异的催化稳定性和结构稳定性.† Supported by the National Natural Science Foundation ofChina(No.41571464) and the Natural Science Foundation of Liaoning Province, China(No.201602467).【相关文献】[1] Schrauzer G. N., Guth T. D., J. Am. Chem. Soc., 1977, 99(22), 7189—7193[2] Ranjit K. T., Varadarajan T. K., Viswanathan B., J. Photochem. Photobiol. A: Chem., 1996, 96(1—3), 181—185[3] Rusina O., Linnik O., Eremenko A., Kisch H., Chem. Eur. J., 2003, 9(2), 561—565[4] Hoshino K., Kuchii R., Ogawa T., Appl. Catal. B: Environ., 2008, 79(1), 81—88[5] Zhang J., The Quantum Efficiency and Mechanism of Nitrogen Photofixation, Zhejiang University, Hangzhou, 2013(张静. 光催化固氮光量子效率及机理研究, 杭州: 浙江大学, 2013) [6] Wang X. C., Maeda K., Thomas A., Takanabe K., Xin G., Carlsson J. M., Domen K., Antonietti M., Nat. Mater., 2008, 8(1), 76—80[7] Liang R. Y., Xu D. D., Cha W. Y., Qi J. Z., Huang L. H., Chem. J. Chinese Universities,2016, 37(11), 1953—1959(梁瑞钰, 徐冬冬, 查文莹, 齐楫真, 黄浪欢. 高等学校化学学报, 2016, 37(11), 1953—1959)[8] Cao Y. H., Tong Y. F., Zhang J., . Li F. Y., Fan Z. P., Bai J., Mao W., Hu S. Z., Chem. J. Chinese Universities, 2016, 37(7), 1357—1363(曹宇辉, 佟宇飞, 张健, 李法云, 范志平, 白金, 毛微, 胡绍争. 高等学校化学学报, 2016, 37(7), 1357—1363)[9] Jun Y. S., Hong W. H., Antonietti M., Thomas A., Adv. Mater., 2009, 21(42), 4270—4274[10] Yu Y. Z., Zhou Q., Wang J. G., Chem. Commun., 2016, 52(72), 3396—3399[11] Bojdys M. J., Muller J., Antonietti M., Thomas A., Chem. Eur. J., 2008, 14(27), 8177—8182[12] Wirnhier E., Doblinger M., Gunzelmann D., Senker J., Lotsch B. V., Schnick W., Chem. Eur. J., 2011, 17(11), 3213—3221[13] Zhao J. N., Ma L., Wang H. Y., Zhao Y. F., Zhang J., Hu S. Z., Appl. Surf. Sci., 2015, 332(1), 625—630[14] Dong G. H., Ho W. K., Wang C. Y., J. Mater. Chem. A, 2015, 3(46), 23435—23441[15] Li H., Shang J., Ai Z. H., Zhang L. Z., J. Am. Chem. Soc., 2015, 137(19), 6393—6399[16] Hu S. Z., Chen X., Li Q., Zhao Y. F., Mao W., Catal. Sci. Technol., 2016, 6(15), 5884—5890[17] Tanabe Y., Nishibayashi Y., Coord. Chem. Rev., 2013, 257(17/18), 2551—2565[18] Jia H. P., Quadrelli E. A., Chem. Soc. Rev., 2014, 43(2), 547—564[19] Sivasankar C., Baskaran S., Tamizmani M., Ramakrishna K., J. Organomet. Chem., 2014, 752(5), 44—58[20] Zhao W. R., Zhang J., Zhu X., Zhang M., Tang J., Tan M., Wang Y., Appl. Catal. B: Environ., 2014, 144(1), 468—477[21] Bojdys M. J., Muller J., Antonietti M., Thomas A., Chem. Eur. J., 2008, 14(27), 8177—8182[22] Wirnhier E., Doblinger M., Gunzelmann D., Senker J., Lotsch B. V., Schnick W., Chem. Eur. J., 2011, 17(11), 3213—3221[23] Zhao J. N., Ma L., Wang H. Y., Zhao Y. F., Zhang J., Hu S. Z., Appl. Surf. Sci., 2015, 332(1), 625—630[24] Wang Y., Wang X. C., Antonietti M., Angew. Chem. Int. Ed., 2012, 51(1), 68—89[25] Wang X. C., Chen X. F., Thomas A., Fu X. Z., Antonietti M., Adv. Mater., 2009, 21(16), 1609—1612[26] Ge L., Han C., Appl. Catal. B: Environ., 2012, 117/118(1), 268—274[27] Lei W., Portehault D., Dimova R., Antoniettit M., J. Am. Chem. Soc., 2011, 133(18), 7121—7127[28] Zhang Y. W., Liu J. H., Wu G., Chen W., Nanoscale, 2012, 4(17), 5300—5303[29] Cheng Y., Pan J., Saunders M., Yao S., Shen P. K., Wang H. T., Jiang S. P., RSC Adv., 2016, 6(56), 51356—51366[30] Ma J. Y., Yin L. W., Ge T. R., Cryst. Eng. Comm., 2015, 17(48), 9336—9347[31] Yu Y., Gao W. Y., Shen Z. X., Zheng Q., Wu H., Wang X., Song W. G., Ding K. J., J. Mater. Chem. A, 2015, 3(1), 16633—16641[32] Ma X. G., Lv Y. H., Xu J., Liu Y. F., Zhang R. Q., Zhu Y. F., J. Phys. Chem. C, 2012, 116(44), 23485—23493。

银参比电极,简称Ag/AgCl电极,是一种常用的金属-金属氯化物电极。
它由银电极和银-氯化银复合物构成。
银参比电极工作原理是利用银电极和银-氯化银复合物之间存在的电动势差来测量电位。
其中,银电极是电子传递体,而银-氯化银复合物是电子传递阻挡物。
银参比电极具有较高的稳定性和线性度,并且在很多pH值范围内都可以使用。
在生物医学和环境科学中广泛用于pH值测量,也在其他领域如分析化学,电化学等领域被广泛使用。
此外,银参比电极还有一些优点,如:
1.高稳定性:银参比电极具有高稳定性,可以在长时间内维持稳定的电动势差。
2.高灵敏度:银参比电极具有高灵敏度,能够精确地测量微弱的电动势差。
3.宽pH范围适用性:银参比电极可以在较宽的pH范围内使用,适用于各种不同环境
的测量。
4.经济实用:银参比电极的制造成本相对较低,是一种经济实用的电极。
5.简单易用:银参比电极的使用简单,不需要高精度的校准和维护,易于使用。
需要注意的是,银参比电极在使用过程中,需要维护和保护,如果没有正确的维护和保护,电极的性能可能会受到影响。


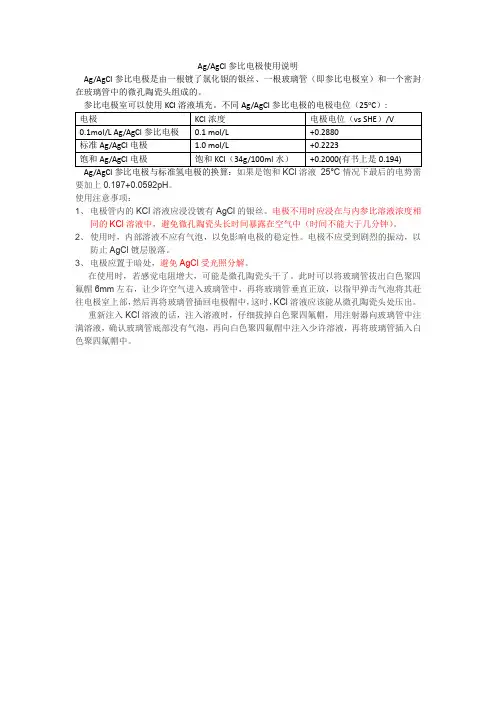
agagcl参比电极电位一、agagcl参比电极电位的定义与作用agagcl参比电极是一种常用的参比电极,其电位被定义为0.1976V (25℃,1mol/L KCl溶液)。
在电化学实验中,agagcl参比电极常用于测量其他电极的电位,通过与待测电极进行比较,可以确定待测电极的电位。
agagcl参比电极的电位稳定性较好,使用方便,因此被广泛应用于各种电化学测量和实验中。
二、agagcl参比电极的构成和原理agagcl参比电极由三个主要部分组成:银电极、银氯化物(AgCl)和KCl溶液。
银电极是主体部分,由纯银制成;银氯化物是电极表面的一层薄膜,由银和氯离子反应生成;KCl溶液则是用于维持电极内部的离子浓度平衡。
根据Nernst方程,agagcl参比电极的电位与溶液中的氯离子浓度相关,因此通过调节KCl溶液中的浓度可以改变参比电极的电位。
三、agagcl参比电极的使用注意事项1. 参比电极的电位与温度密切相关,通常需要进行温度补偿。
在实际操作中,可以使用温度传感器测量温度,并结合温度校正公式对电位进行修正。
2. 参比电极的维护也非常重要。
在使用过程中,应定期清洗和校准参比电极,以确保其电位的准确性和稳定性。
3. 在实验过程中,参比电极的连接方式也需要注意。
通常情况下,参比电极应与待测电极通过盐桥相连,以保持两个电极之间的电位平衡。
4. 参比电极的选择也需根据实际需求进行。
除了agagcl参比电极,还有其他种类的参比电极可供选择,如银银离子电极(Ag/Ag+)和饱和甘汞电极(Hg/Hg2+)。
不同的参比电极适用于不同的实验条件和测量要求。
四、agagcl参比电极在电化学实验中的应用agagcl参比电极广泛应用于电化学测量、电解和电池等实验中。
例如,在PH测量中,可以使用agagcl参比电极与玻璃电极相结合,通过测量电位差来确定溶液的PH值。
在电解实验中,参比电极可用于测量电解池中产生的电位差,从而计算出电解反应的标准电极电位。

agagcl电极用途AG/AgCl电极是一种常用的电化学电极,常用于电化学传感器、电解池、电池以及其他电化学实验中。
它由两个部分组成,一个是银(Ag)电极,另一个是银氯化物(AgCl)膜。
下面将详细介绍AG/AgCl电极的结构、原理以及具体的应用。
AG/AgCl电极的结构由下面的部分组成:1.Ag电极:电极常用银材料制成,因其导电性能好、稳定性高而广泛使用。
2.AgCl膜:AgCl是一种稳定的不溶于水的化合物,它的存在稳定了电极的电位,同时也起到保护Ag电极的作用。
AG/AgCl电极的工作原理是基于电极的离子交换。
当AG/AgCl电极置于电解质中时,电解质中的氯离子(Cl^-)会与Ag电极表面的银离子(Ag^+)发生反应,生成AgCl沉淀。
这个反应会使得AgCl膜的厚度不断增加,从而稳定电极的电位。
AG/AgCl电极的应用非常广泛,以下是几个常见的应用领域:1.电化学传感器:AG/AgCl电极常用于电化学传感器中,用于检测和测量环境中的各种离子,如氯离子、氟离子、铁离子等。
AG/AgCl电极的离子选择性能好,可以与特定的离子发生反应,从而实现离子的测量。
2.酸碱中和电位电极:AG/AgCl电极可以用作酸碱滴定中的感应电极,根据滴定过程中电位的变化来判断酸碱中和的终点。
在酸碱滴定中,当试样液中的酸碱物质被完全滴加到试剂中时,电位会突然发生变化,这时可以通过检测AG/AgCl电极的电位来确定滴定的终点。
3.电解池:AG/AgCl电极常用于电解池中,用于提供电解池中的氧化还原反应的电势。
在电解池中,AG/AgCl电极的电位被控制在一个恒定的值上,使得电解反应能够顺利进行。
4.电池:AG/AgCl电极也广泛应用于电池中,如氯化银电池。
在氯化银电池中,AG/AgCl电极是正极,根据反应AgCl=Ag+Cl-,Cl-离子释放电子给Ag离子,从而产生电流。
除了以上应用领域,AG/AgCl电极还可以用于测量溶液的电位、测量电解质浓度、研究电化学反应动力学等。

agagcl电极用途Ag/AgCl电极是一种常见的电化学电极,由银(Ag)和氯化银(AgCl)组成。
它在许多电化学应用中被广泛使用,包括电化学反应的研究、电化学分析和工业中的电化学处理。
这篇文章将详细介绍Ag/AgCl电极的用途和原理,并讨论它的优点和限制。
首先,Ag/AgCl电极在电化学反应的研究中起着重要的作用。
由于Ag/AgCl电极对一般溶液中的电化学反应不具有催化作用,因此可以被用作参考电极。
参考电极是用来提供一个已知电位的电极,作为与研究电化学反应电位的比较基准。
Ag/AgCl电极通常在离子强度较高的溶液中具有稳定的电位,因此被广泛应用于电化学研究中。
其次,Ag/AgCl电极广泛应用于电化学分析。
它被用作配位电化学法、阳极溶出法和电位滴定法等电化学分析技术中的工作电极或参考电极。
Ag/AgCl电极可以在大多数水溶液中提供稳定的电位,并且具有较好的重现性和准确性。
它的应用范围涉及环境监测、食品安全、药物分析和生物传感等领域。
此外,Ag/AgCl电极在工业中的电化学处理过程中也具有重要作用。
例如,在电解过程中,Ag/AgCl电极可用作阴极,在电解质溶液中被电极化,发生还原反应。
这种反应有助于金属的电镀、电解水和电解制备其他化学品。
在环保领域,Ag/AgCl电极还广泛应用于废水处理、电化学除垢和电化学脱盐等过程中。
Ag/AgCl电极的工作原理基于银离子(Ag+)在氯离子(Cl-)的存在下,发生还原反应生成Ag和AgCl。
Ag/AgCl电极具有良好的电化学稳定性和可逆性。
氯离子的存在使得空气中的氧气不能直接与Ag反应,抑制了银电极的腐蚀。
氯化银的沉淀形成了一个固定的薄膜,保护了银电极免受溶液中其他物质的干扰。
Ag/AgCl电极具有许多优点。
首先,它的制备简单且成本低廉。
其次,它的电化学稳定性高,可用于长时间的电化学实验。
此外,Ag/AgCl电极的电位与氯离子的浓度有关,可以通过调节氯离子浓度来控制电极的电位,提高电位的灵敏度和控制能力。

氯化银参比电极1 氯化银参比电极介绍氯化银参比电极是由金属银与氯化物共同制成的电极,它是当前应用最为广泛的一种参比电极,能够在无氧环境下进行电流测量。
从技术参数上来说,氯化银参比电极是具有高精确度、高稳定性和低噪音的,它的消耗功耗非常低,特点是可以运作在湿润状况下,而且它的反应速度比较快,可以十分准确的测量电流。
2 氯化银参比电极的作用氯化银参比电极的应用十分广泛,它常用于电化学反应中,用于测量反应中的电流,可以隔离测量环境和反应体系内的外界电位,以达到准确测量电流的目的。
此外,氯化银参比电极还可以应用于分析和检测化学物质,常用于实验室;另外,它还常用于太阳能电池反应,用于检测和诊断太阳能电池,有利于开发太阳能电池的性能和可靠性;氯化银参比电极还用于离子电导检测及其它水质检测方面。
3 氯化银参比电极的结构氯化银参比电极是金属银与海藻酸钠同时结合制成,其结构由内外层结构组成,外层是海藻酸钠,中层是电极极材,内层是金属银,银的厚度以及它的孔隙率影响着氯化银参比电极的精度,不同的厚度有着不同的精度要求,电极的极材也有一定的导磁特性,对电极的精度有着一定的影响。
4 氯化银参比电极的优缺点氯化银参比电极有着众多的优势和缺点。
它的优势是:它的精度和可靠性较高,保持电位稳定,反应速度快,消耗功耗低,耐湿性也非常好;缺点是:氯化银参比电极本身存在着一定的上游慢性效应,它会影响该参比电极在不同状况下的精度,同时,由于受外界因素影响,可能会增加参比电极的干扰电平,影响其精度和测量的准确性,因此外部的干扰因素应当加以限制到最小。
5 氯化银参比电极的正确使用步骤氯化银参比电极的正确使用步骤:第一步,清洗电极,确保银表面没有附着物;第二步,测量仪器设置,根据实际应用环境和测量需要,调整仪器参数并确保仪器精度;第三步,连接电极,将电极与仪器连接起来,确保两者之间的接触可靠;第四步,测量参比电极,根据实际需要确定测量时间,以确认电极的变化率,从而得出准确的电流测量结果。


理想的参比电极必须具备如下性质:
1.电极表面的电极反应必须可逆的,电解液中的某化学物质必须服从能斯特平衡电位
方程式(也成为Nernst效应)
2.电极电位随时间的漂移小
3.流过微小的电流时,电极电位能迅速恢复原状(不产生滞后现象)
4.像Ag/AgCL那样的电极,要求固相不溶于电解液
5.当温度发生变化时,一定的温度能响应有一定的电位(没有温度的滞后)
现在经常使用的参比电极有下面三种:
(1)金属相或者溶解的化合物分别与其离子组成平衡体系
H+/H2(Pt)
[Fe(Cp)2]+/Fe(Cp)2(Cp为茂基)
Ag+/Ag
汞齐型 M+/M(Hg)
(2)金属与该金属难熔化何物电离出少量离子组成的平衡体系
AgCI/Ag
Hg2CI2
Hg2SO4/Hg
HgO/Hg
(3)其他体系
玻璃电极
离子选择性电极。

ag agcl电极用途AG/AgCl电极是一种重要的电化学传感器,在许多领域中具有广泛的应用。
下面将介绍AG/AgCl电极的用途以及其工作原理。
AG/AgCl电极最常见的应用是在电化学分析中,用于测量电化学反应的电势或电流。
它可以用于测量溶液中的pH值、氧气浓度、离子浓度等参数。
由于其优异的稳定性和可重复性,AG/AgCl电极常常被用作参比电极、工作电极或计量电极,以保证测量结果的准确性和可靠性。
在环境监测中,AG/AgCl电极可以用于测量水体中的重金属离子、氨氮、硝酸根、温度等参数,从而评估水质的污染程度和环境的健康状况。
此外,AG/AgCl电极还可以用于土壤和气象监测领域,用于测量土壤中的离子浓度和土壤电导率,或测量大气中的气象参数如气温、湿度等。
在医学领域,AG/AgCl电极常用于测量生物体内的生理参数,如血液中的pH值、离子浓度和氧气浓度,从而帮助医生诊断病症和监测病情。
此外,AG/AgCl电极也可以用于心脏电生理学研究中,记录心电图信号,检测心脏功能异常。
在食品工业中,AG/AgCl电极可以应用于食品的质量控制和监测。
例如,它可以用于测量食品中的离子浓度、pH值和氧气浓度,以评估食品的新鲜度、酸碱度和抗氧化能力。
此外,AG/AgCl电极也广泛应用于材料科学、能源储存和生物技术等领域。
在材料科学中,它可以用于评估金属、合金和塑料等材料的腐蚀性能。
在能源储存中,AG/AgCl电极可用于构建锂离子电池、燃料电池和超级电容器等电化学装置。
在生物技术中,AG/AgCl电极可以应用于DNA测序、蛋白质电泳和细胞膜电位测量等实验中。
AG/AgCl电极的工作原理是基于银和氯化银之间的反应。
银电极在电解液中会与氯离子反应生成氯化银沉淀,从而形成银/氯化银电极。
当电极与溶液接触时,溶液中的离子将在电极上发生氧化还原反应,导致电极出现电位差或电流,而这些变化可以被测量和记录下来。
银/氯化银电极可通过连接到外部测量仪器来读取和分析电位或电流信号。
银-氯化银参比电极使用温度银-氯化银参比电极是一种常用的电化学测试电极,广泛应用于电化学分析和电化学传感器中。
它由银(Ag)和氯化银(AgCl)两个部分组成,其中Ag是电极的导电部分,AgCl是可溶性盐的固体部分。
银-氯化银参比电极主要用于测量电动势和电位的稳定性。
它具有稳定、反应迅速、易于校准和使用的优点,被广泛应用于化学、生物、环境和材料等领域。
银-氯化银参比电极的工作原理是基于银和氯离子之间的氧化还原反应。
在Ag中,银原子会失去电子,形成Ag+离子,而氯离子则与银离子结合,生成AgCl固体。
这种氧化还原反应能够保持电极的电势稳定性。
银-氯化银参比电极在使用过程中需要注意一些因素,其中温度是一个重要的考虑因素。
温度的变化会影响银-氯化银参比电极的电势。
一般来说,参比电极的电势与温度有直接的关系。
随着温度的升高,银-氯化银参比电极的电势也会随之增加。
为了确保银-氯化银参比电极在不同温度下的准确性和稳定性,需要进行温度校正。
通常情况下,可以通过使用内标参比电极或温度补偿电路来进行校正。
内标参比电极是一种具有与银-氯化银参比电极相似特性的电极,在不同温度下具有相同的电势变化。
通过与内标参比电极同时进行测量,可以根据其电位差来进行温度校正。
此外,还可以使用温度补偿电路对银-氯化银参比电极的温度变化进行补偿。
该电路可以根据温度的变化来控制电势的稳定性。
通过将温度传感器与电极连接,可以实时监测温度变化,并根据温度变化来调整电极的电位,以保持电势的稳定性。
总之,银-氯化银参比电极是一种重要的电化学测试电极,在不同温度下具有良好的电势稳定性。
通过合适的温度校正和温度补偿措施,可以确保电极在实际应用中的准确性和稳定性。
文章编号:1009-671X(2005)06-0062-02Ag/AgCl 高温参比电极的制备张 清1,李 萍2,白真权3(1.河南科技大学材料科学与工程学院,河南洛阳471003;2.洛阳工业高等专科学校建筑工程系,河南洛阳471003;3.中国石油天然气集团公司石油管材研究所,陕西西安710065)摘 要:在对美国Cortest 公司设计的A g/A gCl 参比电极进行改进的基础上,采用电解法制备了一种内置式Ag /AgCl 参比电极,对其进行了性能测试,并用其对油管钢N 80的高温高压腐蚀行为多次进行了测试研究.结果表明,该参比电极具有重现性好、可逆性好、响应时间短、内阻小等优良性能,且在高温高压条件下工作性能稳定,测试曲线理想,可用于高温高压电化学研究.关键词:Ag/AgCl 参比电极;电解;制备中图分类号:T G145 文献标识码:A收稿日期:2004-12-28.项目资助:中国石油天然气集团公司石油管材研究所基金资助项目(23-524);河南科技大学科研基金资助项目(200101).作者简介:张 清(1974-),男,硕士研究生,主要研究方向:油田腐蚀与防护.Preparation of an elevated temperature Ag/AgCl reference electrodeZHANG Qing 1,LI Ping 2,BAI Zhen_quan 3(1.School of M ater ials Science and Engineer ing ,Henan U niversity of Science and T echnology ,Luoyang 471003,China; 2.Depar t -ment of Architectur e Eng ineering,Luoyang College of T echnology ,Luoy ang 471003,China; 3.T ubular G oo ds Research Center,China N ational Petro leum Corporation,Xi an 710065,China)Abstract:On the basis of the improvement of Ag/AgCl reference electrode of Cortest Company in America,the internal Ag/Ag Cl reference electrodes w ere prepared by means of electrolyzing.T heir features w ere tested and the corrosion behavior of tubular steel N80under hig h temperature and high pressure w ere studied w ith them.T he results show that it has such outstanding features as good reproducibility and reversibility ,short response time and sm all resistance.The new electrodes worked stably and ideal test curves were obtained under high tem -perature and high pressure,and c ould be used in electrochemic al research under high temperature and high pre ssure.Keywords:Ag/AgCl reference electrode;electrolyzing;preparation 为了研究油气钻采设备及石油化工设备在高温高压下的电化学腐蚀机理,模拟油气田高温高压环境下的电化学测试工作愈来愈引起腐蚀科技工作者的重视.高温高压下的电化学测试,一个主要的问题是高温参比电极的制备,目前国内尚未较好地解决这个问题.甘汞电极在室温下使用最广泛,但在温度高于60 时性能不稳定;氢电极在理论上已得到充分研究,但其应用受到很多限制,使用起来很不方便;Ag /AgCl 电极具有制备简单、稳定性好、没有滞后、使用方便等优点,是高温高压电化学研究最理想的参比电极[1].中国石油天然气集团公司石油管材研究所引进了美国Cortest 公司设计的内置式Ag/Ag Cl 参比电极,它由一个很长的聚四氟乙烯管作为盐桥,通过一个不等温的电解液桥与高温区域连通,由于电解液桥温度梯度的存在,所测电位包含了一个由热扩散引起的成分,从而导致在高温高压釜中进行模拟测试时,所得的测试结果稳定性差.假如这一成分不随时间改变,而是一常数,那么就如M ckie [2]和Danie-lson [3]所述,所测电位很容易转化为氢标电位.本文对这一参比电极系统进行改进,成功地制备了Ag/AgCl 高温参比电极,为建立稳定完善的高温高压电化学测试系统奠定了基础.1 A g/AgCl 参比电极的制备1.1 活性元件的制备Ag/AgCl 电极的制备一般有3种方法:直接氯化法、电解氯化法和热分解氯化法.采用电解法制取第32卷第6期 应 用 科 技 V ol.32, .62005年6月 AppliedScience and Technology Jun.2005的Ag/Ag Cl电极对光的敏感性小,不易分解,常用来制备作为内置式参比电极的Ag/Ag Cl电极.本文采用电解法在银表面制备氯化银薄膜,步骤如下:1)对纯度为99.99%的银丝进行表面预处理,包括除去表面硫化物、除油、活化和清洗等;2)以铂电极作为辅助电极与电源负极连接,处理好的银丝与电源正极连接;3)把银丝与铂电极置于25 下1N HCl溶液中,通4mA直流电3h,氯化过程中需不断摇动银丝;4)电解氯化结束后,检验氯化效果是否合格:银丝表面应为灰色致密的AgCl膜层,均匀而无明显的斑点;在0.1N KCl溶液中与饱和甘汞电极的电位差至少应为35mV,否则应重新氯化;5)按以上方法制备3个Ag/AgCl电极,并将所有电极连接在一起,室温下置于0.1N KCl溶液中;6)浸渍24~48h后,使用高阻抗的PZ286型数字电压表测量任意一对电极之间的电位差,应小于1mV;如果差值太大,将两者短路后再置于0 1N KCl溶液中,24~48h后再测其差值,如果仍大于1m V,需磨去氯化膜,重新氯化.1.2 电极内室的填充Ag/Ag Cl参比电极内室设计为双连接形式,两端均有塞子,一端为聚四氟乙烯塞子,另一端为氧化锆陶瓷隔膜,从一端到另一端依次为聚四氟乙烯塞子、饱和KCl溶液、尼龙绳、氧化锆陶瓷隔膜、饱和KCl溶液、氧化锆陶瓷隔膜,Ag/AgCl电极与聚四氟乙烯塞子连接在一起.该结构可有效地避免活性元件Ag/Ag Cl受到污染.填充电极内室时,应首先移去两端的塞子,利用长针管小心填充内室,排除气泡后,将两端的塞子塞回,确保与聚四氟乙烯管紧密接触.电极内室的填充应注意以下3点:1)电解液必须经过除氧处理,因为在酸性溶液中Ag/AgCl电极对痕量的氧是非常敏感的;2)电解液为饱和KCl溶液,可有效地避免低浓度KCl溶液容易出现的不稳定现象;3)AgCl在饱和KCl溶液中的溶解度很大,是在1N KCl溶液中的65倍,因此需在电极内室的KCl 溶液中预先加入少量AgCl粉末,使其达到饱和,避免电极氯化层的溶解,从而保证其性能.2 Ag/AgCl参比电极的性能制得的Ag/Ag Cl电极是否可以作为参比电极,除了测量该电极与饱和甘汞电极之间的电位差外,还应测试该电极的其他一些性能,如重现性、可逆性、响应时间等,从而对参比电极进行综合评价.经测试,室温下该电极与饱和甘汞电极之间的电位差为44mV,多次测量基本一致,说明该电极的稳定性与重现性好;施加+10m V电压进行循环微极化时,正反向曲线几乎完全重合,而且线性度非常好,说明该电极具有很好的可逆性;在室温下插入0.1N KCl溶液中,5s后即可达到稳定,从0.1N KCl溶液中转到1N KCl溶液或饱和KCl溶液中,达到稳定的时间也不超过10s,说明该电极的响应时间极短;该电极内阻不到1000 ,内阻相当小.用该电极在高温高压釜中对油管钢N80的高温高压腐蚀行为多次进行极化曲线和交流阻抗测试,结果表明,该电极在高温高压条件下工作性能稳定,测试曲线理想,可用于高温高压电化学研究[4~6].3 结 论1)在对美国Cortest公司设计的Ag/Ag Cl参比电极进行改进的基础上,选用高纯度银丝,经过严格处理,采用电解法成功制得Ag/AgCl电极.2)性能测试结果表明,制得的Ag/AgCl高温参比电极具有重现性好、可逆性好、响应时间短、内阻小等优良性能.3)用制得的Ag/Ag Cl高温参比电极对油管钢N80的高温高压腐蚀行为多次进行了测试,结果表明,该参比电极在高温高压条件下工作性能稳定,测试曲线理想,可用于高温高压电化学研究.参考文献:[1]金兆法,刘华堂,周锦义.Ag/AgCl高温参比电极的试制[J].中国腐蚀与防护学报,1981,1(2):53-56.[2]M CIK E A S.Hig h temperature high pr essure electr o-chemistry in aqueous solutions[M].Houston:NA CE, 1979.[3]DA NI EL SO N M J.Application of linear polarization tech-nique to the measur ement of corrosion rates in simulated geother mal brines[R].Battelle M emorial Institute,U.S.Department of Energ y,1980.[4]周计明.油管钢在含CO2/H2S高温高压水介质中的腐蚀行为及防护技术的作用[D].西安:西北工业大学, 2002.[5]任呈强.N80油管钢在含CO2/H2S高温高压两相介质中的电化学行为及缓蚀机理研究[D].西安:西北工业大学,2003.[6]张 清.CO2/H2S共存条件下的油管钢腐蚀规律研究[D].洛阳:河南科技大学,2004.[责任编辑:李玲珠]63第6期 张 清,等:Ag/AgCl高温参比电极的制备。
氯化银电极的参比电极氯化银电极的参比电极氯化银电极,也称为Ag/AgCl电极,是一种重要的参比电极。
它通常用于电化学实验和分析中,以确保准确的电位测量。
在分析化学、环境分析、生物化学和药物学等领域中,氯化银电极被广泛使用。
因此,深入了解氯化银电极的性质和特点以及其在实验中的应用是十分必要的。
1. 氯化银电极的构成氯化银电极基本由两部分组成:银电极和氯化银盐。
银电极是一根纯银棒或线,常常涂上一层或几层银氯化物。
该电极的原理是基于Ag/AgCl的半反应形成的,其中Ag为远离溶液,AgCl浸泡在溶液中。
当溶液中具有电活性物种,例如NaCl、KCl、AgNO3等时,AgCl不断地与离子溶解并生成Ag+和Cl-离子。
2. 氯化银电极的特点1)稳定性高:氯化银电极使用广泛的原因之一是其具有极高的稳定性。
它的稳定性非常高,即使在长期使用过程中也很难发生变色或腐蚀现象。
这使得氯化银电极成为了做各种实验的首选参比电极。
2)响应速度快:氯化银电极是反应速度非常快的电极之一。
它能够在很短的时间内响应并产生电势差,这使其在多种实验中得到了广泛应用。
3)易于制备:氯化银电极的制备非常简单,只需要将银棒浸泡在氯化银溶液中,待其包覆在一层银氯化物层后即可使用。
其制备过程简单,成本较低,易于推广使用。
3. 氯化银电极的应用1)电化学测量:氯化银电极通常被用于电化学实验中,以测量反应过程中的电势变化。
电势测量是分析化学、环境分析和药物学等学科中的一个重要工具,在分析分子相互作用和判断物质组成和结构等方面起着关键作用。
2)生物化学和医学研究:由于氯化银电极有较高的响应速度和稳定性,且能承受生物体系中的高盐度、氧气和氨气浓度,因此它在生物化学和医学研究中得到了广泛应用。
例如,在生物反应器中测量溶液pH值、蛋白质电位、离子浓度等方面,氯化银电极都发挥了重要作用。
3)环境分析:氯化银电极被广泛应用于环境分析领域。
例如,在污染物检测中,它可以测量水流中的离子浓度,以判断水质是否达到特定标准。
agagcl电极用途Ag/AgCl电极是一种常见的参比电极,被广泛用于电化学分析和电化学实验中。
Ag/AgCl电极由银电极和氯化银组成,具有稳定性好、维护简单和成本低廉等优点。
以下是Ag/AgCl电极的用途的详细介绍。
1.电化学分析:Ag/AgCl电极被广泛用于电化学分析中,如电位滴定、离子选择性电极测定、电位法和电流法等。
在这些分析中,Ag/AgCl电极作为参比电极,提供稳定的参照电位,使得被测物质的电位变化能够精确地测定。
2.pH测量:Ag/AgCl电极也常用于pH测量中,作为玻璃电极的参比电极。
pH是描述溶液酸碱性的重要指标,Ag/AgCl电极的稳定参比电位使得pH测量结果更准确可靠。
3.电解池:在电解过程中,Ag/AgCl电极常用作阳极或阴极,用于传递电流和反应电子。
同时,Ag/AgCl电极的稳定性和成本效益使其成为许多电解池中的理想选择。
4.腐蚀研究:Ag/AgCl电极可用于研究金属和合金在腐蚀环境中的电化学行为。
通过测量电位变化,可以评估材料的腐蚀倾向和腐蚀速率,进而指导材料腐蚀控制和保护的方法。
5.储能装置:Ag/AgCl电极也被用于储能装置中,如锌银电池和镍银电池等。
在这些装置中,Ag/AgCl电极起到了催化反应和电极反应传递电子的作用,使电池能够高效地存储和释放能量。
6.生物传感器:Ag/AgCl电极还被广泛应用于生物传感器中,用于检测和测量生物分子的电化学信号。
通过将特定的生物分子与Ag/AgCl电极表面接触,例如DNA、蛋白质和酶等,可以产生特定的电化学反应,进而实现生物分析和生物检测等应用。
7.医学应用:Ag/AgCl电极也在医学领域中得到广泛应用,如心电图测量中的电极。
在心电图测量中,Ag/AgCl电极用于记录心脏电信号,提供精确的心电图结果。
总之,Ag/AgCl电极在电化学分析、pH测量、电解池、腐蚀研究、储能装置、生物传感器和医学等领域都有重要的应用。
其优越的性能和广泛的用途使得Ag/AgCl电极成为当前电化学研究和应用中的重要部分。
银/氯化银参比电极
产
品
说
明
书
河南邦信防腐材料有限公司
2017年6月
银/氯化银参比电极用纯银及其他贵金属材料制作。
氯化银参比电极有稳定性以及好的电化学稳定性,且外壳采用强度塑料,能够在海水、土壤等复杂环境中长时间稳定使用,因此在阴极保护领域及其他相关领域得到了广泛的应用。
实验室内用银/氯化银参比电极图片
海洋环境、土壤用固态耐压银/氯化银参比电极图片
实验室参比电极介绍
氯化银参比电极分为长效氯化银参比电极及便携式氯化银参比电极两种。
河南邦信公司生产的银/氯化银参比电极主要用于海水或含氯离子环境金属结构的电位测量。
此类电极为双液接Ag/AgCl参比电极,不会被极性化,因此可以提供可靠的数据。
电极分固定安装型与便携式两种。
银/氯化银参比电极在含氯水溶液中具有很高的电化学稳定性,测量精度高(小于10mV) ,与铜/硫酸铜电极的电位差约100mV。
银/氯化银参比电极采用了特殊的密封设计,使用深度可达到30米左右。
设计用途
海水、含盐水、含氯污水中电位监测。
Ag/AgCl银/氯化银参比电极规格型号
使用方法:
1、将Ag/AgCl银/氯化银参比电极封头打开,可直接使用;
2、Ag/AgCl银/氯化银参比电极使用后,尽量浸泡在氯化钾溶液中或清水中保存;
注意事项:
1、搬运时小心轻放,严禁提拉导线移动电极;
2、电极埋设前应放置在阴凉干燥处,避免暴晒或雨淋;
3、冬天要做防冻处理;
4、严禁在深度超过30米水下区域使用;
5、其他事项请咨询生产单位。
河南邦信防腐材料有限公司
技术部
2017年6月。