肝豆状核变性
- 格式:pptx
- 大小:4.04 MB
- 文档页数:44


・298・ 士进修杂志2O02年4月第17卷第4
肝豆状核变性的护理
沈发风
(浙江省湖州市中 L,N院神经内科,浙江湖州3131300)
关键词肝豆状核变性 护理 中图分类号:P,575,2 4 文献标识码:A 文章编号:1 ̄02-6975[2002)0*0"z98-Ce
肝豆状核变性又称Wilson氏病(HLD),是一种
铜代谢障碍为特征的常染色体遗传病,其发病率
为0、5/10万~3/10万” 其特点是铜沉积在肝、
脑、肾、角膜等组织引起一系列临床症状 而早期予
有效的驱铜治疗是改善本病预后的关键,并可获得
近似常人的生活质量和寿命。我院1994年6月~
2000年11月共收治肝豆状核变性患者l3例,取得 满意疗效,现将护理体会报告如下
1临床资料 13例中男4例,女9例,年龄11~43岁,平均年
龄l9.3岁。首发症状:以神经精神症状首发5例;
以肝功能损害首发7例;以急性溶血性贫血首发1
例。13例患者眼科裂隙灯下角膜K-F环均阳性,血 清铜蓝蛋白2O~45 mg/L,平均31 mg/L,(正常参考
值210~530 J L)。尿铜0.37~2.44 J ./24 h,平均
1.07 mg/Z4 h,(正常参考值0、02~0.03 mg/24 h)。 其中1例患者其兄长角膜K.F环阳性。另1例其伯
父生前有肝硬化史,余家族史无殊。
2治疗结果及预后
本病的治疗原则是促进体内铜的排泄以及减少
铜的吸收,以建立铜代谢的负平衡。需终生服药
13例患者经应用促铜盐排泄药物(D一青霉胺或
二巯基丙磺酸钠)与阻止肠遭对铜吸收药物(葡萄糖
酸锌)以及其他对症治疗和精心护理,其中7铡患者
临床症状明显缓解。基本获得常人的生活质量,现用
维持量,并定期门诊复查,随访;4例患者症状得到
控制后转上级医院治疗;2例患者已至疾病晚期,疗
效欠佳,后因经费问题自动出院,失访。
3典型病例 例l:患者,女性,l3岁。园“进行性行走不便伴

精品文档
精品文档 肝豆状核变性 ( hepatolenticular degeneration,HLD),又称为Wilson病(Wilson s disease,WD),是一种常染色体隐形遗传的铜代谢缺陷病,发病率仅0.5/ 10 万~3/10 万,其基因定位于13q14.
3,编码1 个P 型ATP 酶,此酶参与铜跨膜转运的代谢过程。目前研究多认为由于WD 基因突变使其功能降低或丧失而导致铜代谢异常,肝合成铜蓝蛋白速度减慢,胆汁排铜明显减少,铜沉积于肝、脑、肾、角膜、血细胞和关节等组织中,引起了相应脏器损害的临床症状。铜蓄积可导致肝细胞坏死、肝纤维化;从坏死的肝细胞释放的大量铜可导致溶血,并逐渐沉积在脑、肾、角膜、骨关节部位,引起多器官受累。不同程度的肝细胞损害,脑退行性病变和角膜边缘有铜盐沉着(K-F环)为其临床特征。早期诊断和治疗可避免严重的不可逆的组织器官损害,常可获得与健康人一样的生活和寿命。
1921年Hall首先分析68例有血缘关系的HLD病人,其中34例至少与另一名患者有血缘关系,且绝大多数是表(堂)亲。因此Hall确信HLD属遗传性疾病,并且指出是由2个不同的隐匿性代谢缺陷的基因传递所致,其传递形式与常染色体隐性遗传相一致。我国调查167例HLD先证者的家系,615位先证者及其同胞中,278人证实为HLD,按Hardy-Weinberg公式计算;(278-167)/(615-167)=0.248。此值与常染色体隐性遗传的预期值0.25极接近,统计学无显著差异(P>0.05),符合常染色体隐性遗传。
肝豆状核变性的临床表现
肝豆状核变性是一种先天代谢缺陷病。其病理生理基础是铜代谢呈正平衡。全身组织内有铜的异常沉积。本病散见于世界各地不同的民族。从遗传学说研究估计本病发病率约为人口的 16万分之一。我国(包括台湾)的不完全统计有220例。大多数在少年或青年期发病,以10~2 5 岁最多,男女发病率相等。幼儿发病多呈急性,在数月或数年内死亡,30岁以后发病多属慢 性型。

综 述
肝豆状核变性药物治疗现状及进展
邓晰明
肝豆状核变性(W ilson s d i sease,W D是一种常染色体隐性遗传的铜代谢障碍性疾病,由于过多的铜沉积于肝脏、基底神经节、肾脏等处,临床表现为肝脏、脑、肾脏等脏器的损害。W D作为少数几种可治疗的遗传性疾病之一,早期、及时、有效的治疗对改善患者的生活质量、降低病死率均有重要意义。
一、W D的治疗原则
由于患者的AT P7B基因突变导致铜代谢障碍,铜在机体内缓慢累积而出现相应症状。针对以上发病机制,目前临床治疗原则主要包括:!减少铜的摄入,主要是坚持低铜饮食及使用减少铜在肠道吸收的药物;∀增加铜的排泄,目前主要应用金属螯合剂与铜离子结合后排出体外;#针对W D表现为暴发性肝衰竭和晚期肝硬变的患者,可以给予肝移植或干细胞移植治疗;∃对症治疗。
二、药物治疗
1.减少铜吸收的药物
锌制剂:常用的有硫酸锌、葡萄糖酸锌和醋酸锌等。目前认为锌制剂的治疗机制有:(1在肠道与铜竞争,可减少肠道对铜的吸收;(2可以诱导肠粘膜内金属巯蛋白的产生,而金属巯蛋白与铜的结合能力较强,使得较多的金属巯蛋白-铜复合体储存于肠道粘膜内而随粘膜脱落从粪便排出;金属巯蛋白还可减轻金属毒物对机体的氧化损伤[1];(3阻止唾液、胃液及肠液中再分泌铜的重吸收,从而达到铜的负平衡[2]。1979年国外报道了3例W D患者口服硫酸锌治疗获得临床改善[3]。国内杨任民等报道应用10%硫酸锌口服10m l/d治疗60例,3~4周后临床症状改善率80%,治疗后第2周开始尿排铜量显著增加,血铜水平下降而血锌增高[4]。鉴于硫酸锌的消化道等不良反应较大,杨任民等在国内首次将葡萄糖酸锌用于W D的治疗,发现大剂量葡萄糖
酸锌治疗组(1.68g/d患者临床症状改善明显,尿排铜增高,血清铜降低,血锌增高,疗效与硫酸锌相同而不良反应小,为治疗W D较为理想的锌制剂[5]。临床应用及动物实验均证明,锌剂治疗确实能够降低肝铜含量,延缓由于铜中毒引起的临床症状及病理变化的发生[6]。国外应用硫酸锌治疗症状前期患者,治疗过程中尿铜量明显增加,治疗3~ 5年患者均未出现临床症状及药物不良反应[7]。甚至部分学者认为,W D临床症状的出现主要与血中游离铜的毒性有关,而与沉积铜和铜蓝蛋白结合的铜无关,因此推荐使用锌制剂代替金属螯合剂治疗W D[1]。
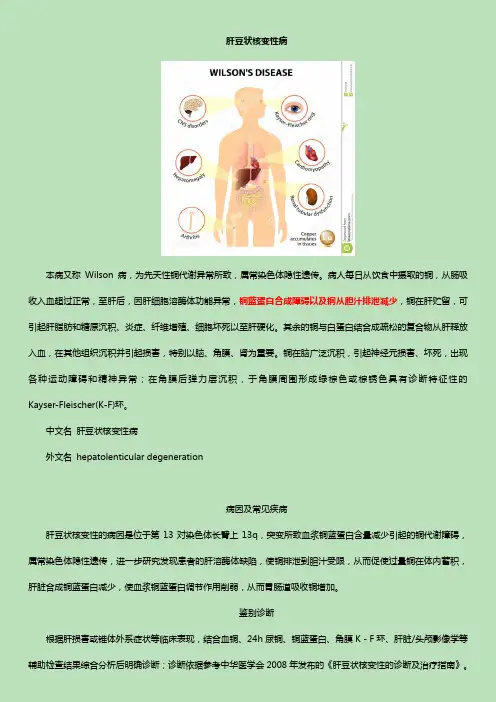
肝豆状核变性病
本病又称Wilson病,为先天性铜代谢异常所致,属常染色体隐性遗传。病人每日从饮食中摄取的铜,从肠吸收入血超过正常,至肝后,因肝细胞溶酶体功能异常,铜蓝蛋白合成障碍以及铜从胆汁排泄减少,铜在肝贮留,可引起肝脂肪和糖原沉积、炎症、纤维增殖、细胞坏死以至肝硬化。其余的铜与白蛋白结合成疏松的复合物从肝释放入血,在其他组织沉积并引起损害,特别以脑、角膜、肾为重要。铜在脑广泛沉积,引起神经元损害、坏死,出现各种运动障碍和精神异常;在角膜后弹力层沉积,于角膜周围形成绿棕色或棕锈色具有诊断特征性的Kayser-Fleischer(K-F)环。
中文名 肝豆状核变性病
外文名 hepatolenticular degeneration
病因及常见疾病
肝豆状核变性的病因是位于第13对染色体长臂上13q,突变所致血浆铜蓝蛋白含量减少引起的铜代谢障碍,属常染色体隐性遗传,进一步研究发现患者的肝溶酶体缺陷,使铜排泄到胆汁受限,从而促使过量铜在体内蓄积,肝脏合成铜蓝蛋白减少,使血浆铜蓝蛋白调节作用削弱,从而胃肠道吸收铜增加。
鉴别诊断
根据肝损害或锥体外系症状等临床表现,结合血铜、24h尿铜、铜蓝蛋白、角膜K-F环、肝脏/头颅影像学等辅助检查结果综合分析后明确诊断;诊断依据参考中华医学会2008年发布的《肝豆状核变性的诊断及治疗指南》。 检查
特征性实验室检查:血浆铜蓝蛋白减少,低于20mg/dl;血清总铜减少。血清直接反应铜增高;尿铜排出增多。肝活检肝细胞含铜量增多。
治疗原则
现如今用于治疗Wilson病的药物包括青霉胺、曲恩汀、四硫钼酸铵及锌剂等。Wilson病一经诊断,则需终身治疗。
治疗主要为防止铜继续在组织累积并将已沉积的铜移去。排铜首选青霉胺,也可用二巯基丙醇(BAL),但疗效较差;其次二盐酸三乙烯四胺。应采用低铜饮食;硫酸化钾在进餐时服用,可减少铜从胃肠道吸收。对肝、脑症状,给予对症治疗。