药物特殊毒性评价
- 格式:ppt
- 大小:764.50 KB
- 文档页数:44



药物安全性评价绪论一、药物安全性评价概述药品:指用于预防、治疗、诊断疾病,有目的地调节人的生理机能并规定有适应症或者功能主治、用法和用量的物质,包括中药材、中药饮片、中成药、化学原料药及其制剂、抗生素、生化药品、放射性药品、血清、疫苗、血液制品和诊断药品等。
(《中华人民共和国药品管理法》)(一)新药研发程序1.探索和筛选:应用常规的药理学方法检测化合物的药理学活性,超过活性标准即可认为有活性;通过早期毒性筛选尽量排除可能有问题的化合物,从而增加测试化合物的数量,减少实验动物的用量和扩大给药剂量的范围。
2.非临床研究:实施符合GLP标准的毒理学安全性评价、生物利用度评价及药代毒代动力学研究。
3.临床研究:Ⅰ期:证明人类对该药物的耐受性并确定其在人体体内的药物代谢动力学特性。
Ⅱ期:确定药物的量效关系。
Ⅲ期:临床药效学试验(全面、多中心、大量患者参与)Ⅰ期、Ⅱ期、Ⅲ期临床试验应符合GCP标准。
4.申请新药注册:《药品管理法》、《药品注册管理办法》5.新药上市:进行药物不良反应监测,并进行符合GCP标准的Ⅳ期临床试验(二)安评的起源最早提出药物安全性评价是缘于20世纪全世界出现了许多严重的药物中毒事件。
1.磺胺酏剂事件(30年代,美国)a.1937年,美国一家公司的主任药师瓦特金斯(HaroldWotkins)为使小儿服用方便,用二甘醇代替酒精做溶媒,配制口服液体制剂,称为磺胺酏剂。
b.未做动物实验,在美国田纳西州的马森吉尔药厂投产后,全部进入市场,用于治疗感染性疾病。
c.到这一年的9~10月间,美国南方一些地方开始发现患肾功能衰竭的病人大量增加,共发现358名病人,死亡107人(其中大多数为儿童),成为上世纪影响最大的药害事件之一。
2.有机锡中毒事件(50年代,法国)a.烷基锡化合物可引起多起中毒。
b.1954年在法国用于治疗皮肤化脓很有效的Stallion药物,这种药物中含有作为不纯物的10%三乙基锡碘化合物,有1000人服用这种药物,有约100人死亡。

保健食品安全性毒理学评价试验的四个阶段和内容发布日期:2021-04-03浏览次数:1605字号:[大中小]毒理试验的四个阶段和内容1第一阶段:急性毒性试验口服急性毒性:LD50,联合急性毒性,一次性最大耐受性受量试验。
二阶段II:遗传毒性试验,30天喂养试验,传统畸试验遗传毒性试验的组合应考虑原核和真核细胞的结合原理,体内试验和体外试验。
来自Ames试验或V79/HGPRT基因突变试验、骨髓细胞微核试验或哺乳动物骨髓细胞染色体畸变试验、4.1.2.3或4.1.2.4试验别各选一项。
2.1基因突变试验:首选鼠伤寒沙门氏菌/哺乳动物微粒体酶试验(Ames试验),其次是V79/HGPRT基因突变试验,必要时可另选其它试验。
2.2骨髓细胞微核试验或哺乳动物骨髓细胞染色体畸变试验。
2.3tk基因突变试验。
2.4小鼠精子畸形分析或睾丸染色体畸变分析。
2.5其它备选遗传毒性试验:显性致死试验、果蝇伴性别隐性致死试验,非程序DNA合成试验。
2.630天喂养试验。
2.7传统致畸试验。
三第三阶段:亚慢性毒性试验-90天喂养试验和繁殖试验殖试验、代谢试验四第四阶段:慢性毒性试验(包括致癌试验)1、药物的毒性试验分为两类,其一特殊毒性试验,包含致癌、致残、致突变,号称三致试验;作此试验的单位必须是国家指定并认可的;一般单位即使做了也不被国家认可!通常一类创新药必须做的。
无论一个药物的药效如何好,只要含有三致试验的特殊毒性(动物若干代繁殖后,查看),该药物将不会允许上市的。
2、其二,一般毒性试验,通常用老鼠或兔子去做,主要是测定半数致死量;若某个药物的半数致死量是有效使用剂量的2倍以上,通常认为是临床使用安全的,若半数致死量比有效剂量略大一点,那就谁也不敢使用了,万一略微超一点量就会导致患者死亡!。

药物安全性评价实验―――急性毒性实验摘要:通过药物安全性评价实验,来测定药物的毒性的强弱,为长期毒性实验或特殊毒性实验提供剂量依据,得到相关新药毒性的信息,本次实验采用急性毒性实验(即LD50的测定),以硫酸链霉素作为安全评价的药物,小白鼠作为实验对象,分别注射不同浓度的原药来观察其小鼠的死亡情况及其死亡时的现象,剖解后的生理状况,然后根据其改良寇氏法代入得到的相关数据来测定出药物的半数致死量,从而来评价硫酸链霉素的毒性大小.[注:本组在做正式实验时负责的浓度为1/6原药浓度在第二组浓度的0.7倍浓度]关键词:急性毒性 LD50 剂量设计现象一目的和意义:1.根据急性毒性实验可以测定药物毒性的强弱2.根据急性毒性实验可以为长期毒性实验或特殊毒性实验的剂量设置提供依据3.根据急性毒性实验可以获取新药毒性反应的相关信息二实验仪器动物与药品药品:硫酸键霉素规格:2ml:0.5g 100万单位 10支器材:灭菌锥形瓶 10ml移液管镊子解剖剪注射器碘酊等实验对象:小白鼠三剂量设计:预实验组1.目的:找到药物的最小致死量和药物的最大致死量的大约范围为正式实验做好数据准备。
2. 药品对象及仪器:药品:磺胺间甲氧嘧啶钠注射液规格:5ml:0.5g器材:灭菌锥形瓶 10ml移液管镊子解剖剪注射器碘酊等实验对象:小白鼠4只3.剂量设置:4.实验内容:4.1 称取小白鼠,将小白鼠称重,同重的做同一标记,放在同一笼子里面,做到雌雄各半,然后采用随机分组的原则进行(本次实验按照体重随机分组)。
每组分4只4.2 配药(即药液的稀释)采用等比稀释的方法进行,分别将原药稀释成原药 1/2 1/4 1/8这四个浓度备用4.3 给药(本次实验采用腹腔注射)4个组分别注射4个不同浓度,给药量按0.2ml/10g的标准进行给药。
4.4 观察给药后连续观察30min(表现在:外观皮毛,活动情况,姿势,精神情况,以及有无舔脚,呼吸困难,体温变化,腹泻,眼睛,肛门,口鼻耳的变化等)记录其情况以及死亡情况,1―――4小时后观察其情况。
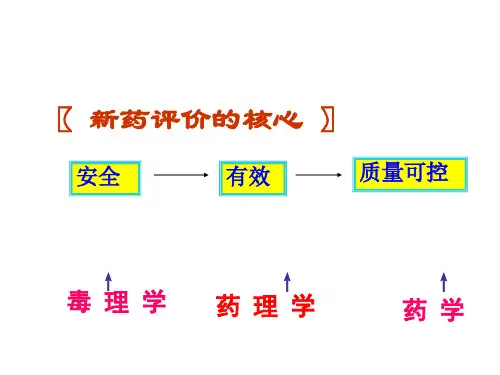




药物毒性的评价在美国,食品和药品管理局(FDA)管理药物的研制.动物研究所得的药理学和毒理学资料(临床前)作为研制中新药(IND)申请的一部分提交FDA.如果这些资料证明该药充分安全的和有效的,人体(临床)研究将分三期进行;从人体研究所获得的资料作为新药申请(NDA)的一部分提交FDA.虽然FDA要在6个月以内对NDA作出答复,但实际上批准新药申请(NDA)常需2~3年时间.药物开发的总时间即从提出IND到最终批准NDA平均为8~9年.动物研究在进行人体研究之前,必须按照FDA制定的法规(良好实验室规范GLP)对药物的动物研究所得的药代动力学,药效动力学和毒理学性质进行评定并形成文件.有两项主要设想要作出:化合物在适宜的实验动物试验中得到的作用可用于人类;为发现可能在人体出现的毒性,对实验动物使用大剂量是一个必需的及有效的方法.由于试验中所用的动物数相对较少,而要检出发生率低的毒性反应,高剂量是必需的.药物的安全性是通过在几种动物身上进行急性,亚急性和慢性毒性试验来确定的.急性毒性试验最初的急性毒性试验是测定致死量(LD50或LD90,分别指实验动物死亡50%或90%所需的药物剂量),中毒症状和症状出现的时间.通常,至少使用三种动物(其中一种为非啮齿类),而且急性毒性常需采用一种以上用药途径来确定.近年来,测定致死量所用动物数已减少,但也相应地降低其精确性.现已认识到全面评估药物对人体的毒性时这种高精确性并非必要,因为仅测定LD50或LD90这一项指标对人没有多少预估价值.除非与除死亡之外的毒性测量的长期试验资料结合起来才有意义.亚慢性毒性试验亚慢性毒性试验至少要在两种动物身上进行,通常每天给予试验药物持续给药时间可长达90天.在每种动物至少应用三个剂量水平,从预期的治疗剂量直到足以引起毒性的高剂量.所使用的给药途径最好与人体试验的途径一致.体格检查及化验检查应在整个观察期内进行.在试验结束时要将动物处死,并作病理检查以确定受累的器官.慢性毒性试验慢性毒性试验至少在两种动物身上进行,其中一种为非啮齿类动物.这种试验通常要长达实验动物一生的时间(即啮齿类2年,非啮齿类2年以上).试验的长短还取决于该药打算在人体应用的时间.要使用三个剂量水平:非毒性的低剂量水平,高于预期治疗量的剂量,及足以产生中毒反应的剂量.而且在整个用药期间,每隔一段时间要进行体格检查和化验检查.一些动物可以定期处死,作大体观察和组织学检查.根据这些试验结果,研究者可以确定药物影响哪些器官,以及该药是否具有潜在的致癌作用.此外,要在大鼠和家兔身上进行广泛的生殖试验以测定在生殖周期中的变化以及有否致畸作用.这些试验和慢性毒性试验可以和人体的初期试验同时进行,特别是该药只打算在人体作短期使用时.体外研究近来,人们对应用体外毒性试验越来越感兴趣,这种试验比动物试验更快捷,具有高的价格-效应比.体外研究的重点是在致突变方面,所采用最广泛的试验方法就是Ames生物检定法.某种化合物若是显示为一种细菌的致突变原就有可能是哺乳动物的致癌物.体外毒性试验并不是要取代动物试验,仅作为在规章制订过程中提供支持的信息.体外毒性试验正被制药公司采用于对那些需要进一步体内试验和药物学开发的特殊化合物.而且,体外研究在药物发展中的作用已有所扩展,体外研究用于预测药物在人体内的代谢途径,这种代谢途径可能不同于实验动物.应用能表达主要人体药物代谢酶的细胞株作体外研究可以有助于预期在人体内产生的新的代谢物,该代谢物在动物试验中可能检不出来.可以提交体外试验资料作为传统的吸收,分布,代谢和排泄研究的一个补充.人体研究在FDA批准上市之前新药的人体研究必须分三期进行(新药批准后所进行的药物一般应用研究和售后调研可看作为第4期).人体研究是必需的,因为据估计≥50%的最常见的不良反应(如抑郁,灼心感,头痛,耳鸣)不能在动物研究中辨认出来.由于有毒药物的效应以及由于无效药物引起的日益增加的严重症状在所有人体研究中都是危险的.因此,研究参加者的权利需要有安全保护措施,如主管机关审查委员会的同意的报告以保护受试者的权益.第一期第一期研究,一种新药首次应用于人,通常在少数人(20~80名)年龄为18~45岁的男性健康受试者身上进行.研究的目的是要确认在人体刚出现毒性时的剂量水平,因而是要测定一个安全的可耐受的剂量.因为这些研究的目的指标是毒性,因此有关的同意的报告是必要条件,而且参加者必须受到具有急救措施的医务人员的密切监护.第一期试验能够进行前,先要制订一个有关临床研究的状况及人员的计划书,该计划书要得到IRB的批准再送交FDA,经FDA批准计划书,并发布试验性新药免税许可,这样才能进入第一期试验.试验开始时,每位受试者接受一次药物剂量,并对药物不良反应进行密切监护,如果没有人发生不良反应,药物剂量逐渐增加,直到事先确定的剂量,或达到一定的血清药物水平,或出现毒性时则中止试验.药物的吸收,代谢和排泄也可以测定.第二期第二期研究在安全性已取得满意的初步证据之后即可开始.这一期是在监督之下对80~100名病人给药,该药是用于治疗或预防预期的疾患或症状的.最好是病人除了预期的疾患之外不应有其他的健康问题.第二期研究通常是以新药与用于预期疾患的典型药(如具有的话)相比较,并以随机化方式的试验.通常这也是对人体长期用药效应的首次观察.第二期研究的目的是要确定新药的最佳剂量-效应范围,并证实新药对预期疾患的效能.也应当对参加者进行不良反应监测.因为受试者样本较大,以前未出现的不良反应此期可以出现.第二期是最关键的,因为从该期所得的资料要作出是否在更大人群中进行更广泛的研究.第三期在第一,第二期研究中,药物的安全性和效能已经取得适当的证据.之后即可进入第三期研究,该期一直持续到新药投放市场供普遍应用时为止.该期研究有许多临床医生参与,他们可以对数百名至数千名病人进行监护.第三期研究的目的是要核实受试药物的效能,以及检测出在第一,二期可能没有发生过的效应.其结果是为主办试验者和FDA能够作出该药物使用的安全性和有效性的决定.关于怎样才是安全和有效,尚无明确的规定,必须要根据正在用药治疗的情况和变换治疗的情况才能作出判断.当充分的资料已经收集并证明可以继续使用这药时,就可提交新药申请书.从最早的以药理学为基础筛选出药物到完成填写新药申请书之间通常需要4年或更长的时间.第四期第四期研究是在药物批准后进行;第四期是继续大样本研究,通常还包括特殊的群体如孕妇,儿童和老人.在药物批准之前在这特殊群体中研究是不道德的(如胎儿接触药物的危险)或不科学的(如引进未知的变数).第四期研究能检出低发生率的不良反应.临床前和临床研究中仅能检出药物不良反应发生频率>1:1000的反应,因而在这方面是相对不敏感的.对许多药物来讲,不良反应发生率1:10000~1:50000,这是符合临床情况的.这只能在新药申请书被批准后新药上市后的售后调研时才能检出.新的治疗效应或中毒反应,其中包括那些在少数病人身上罕见的或不能辨出的长期效应,可以被发现.那些由厂商准备的有关药物安全性和效能的小册子或广告必须由FDA审查和批准.在本期内研究报告必须定期送交FDA,在第一年每3个月一次,第二年每6个月一次,再以后每年一次.这些报告必须包含药物的数量以及邮件单据,标签和广告方面的复印件资料.制造商还必须向FDA报告非预期的副作用,伤害,中毒或过敏反应.这样,FDA可继续履行监察责任,以保证药物上市后的安全性和有效性.。
1.试诉新药临床前安全性评价的内容。
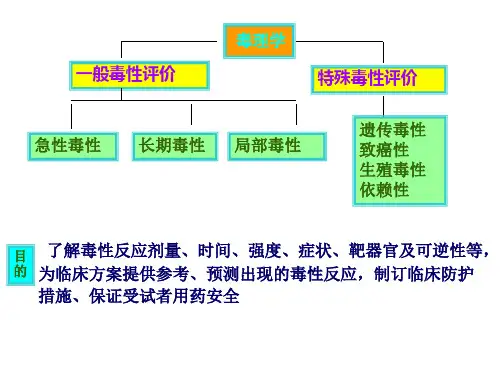
答:新药临床前安全性评价的内容分为两大类,一是一般毒性试验,二是特殊毒性试验。
所谓特殊毒性试验,是指以观察和测定新药能否会引起某种或某些特定的毒性反应为目的而设计的毒性试验,即此类毒件试验观测的毒性指标是明确的。
广义的特殊毒性试验包含的面比较广。
如遗传毒性试验、生殖毒性试验、依赖性毒性试验、过敏性试验、局部刺激性试验、免疫毒性试验、眼毒试验、耳毒试验及致癌试验等,而狭义的特殊毒性主要是指遗传毒性、生殖毒性和致癌性,即-般常说的“三致”试验。
所谓一般毒性试验,是指那些不以观察和测定某种特定的毒性反应为目的而设计的毒性试验,这意味着观测的毒性指标具有广谱性和不确定性的特点、包括生理学、血液学、血液生化及病理形态学等多方面的综合性指标。
如用不同种属的动物及不同给药途径进行的急性毒性试验、反复多次给药的长期毒性试验。
一般药理学试验,是观察新药在一定的剂量条件下,除了主要药效学以外的对机体各系统的影响,国外称为安全性药理试验,理所当然的属于安全性研究的内容,我们国家的新药审评办法,对一般药理试验的要求比较简单,仅要求观察药物对神经、心血管及呼吸系统的影响,而且各系统的试验观察指标也很有限。
另外,除了一般常用的口服、肌内注射、皮下注射、静脉注射途径外,新开发的结药途径及其剂型,其安全性试验的内容,陈了一般性的要求外,尚应根据其具体特点设计能说明问题的毒性试验。
2、试述新药安全性评价的局限性。
答:临床前毒理学评价的基本手段是动物试验,动物实验的第一个局限是而动物由于缺乏第二信号系统,不能象人一样能述说主观感觉性质的毒性反应,如疼痛、腹胀、视物不清、头晕、头昏、耳鸣、疲乏等,这些反应在动物上是难于发现或者是不可能发现的。
另外,由于生物进化上的差异,致使有的反应只出现于人.有的则只出现于动物.人和动物共同都出现的反应仅占一部分。
动物实验的第二个局限是实验动物的数量有限,那些发生率很低的毒性反应,在少量的动物中难以发现。