药物Letermovir(莱特莫韦)合成检索总结报告
- 格式:pdf
- 大小:766.57 KB
- 文档页数:9
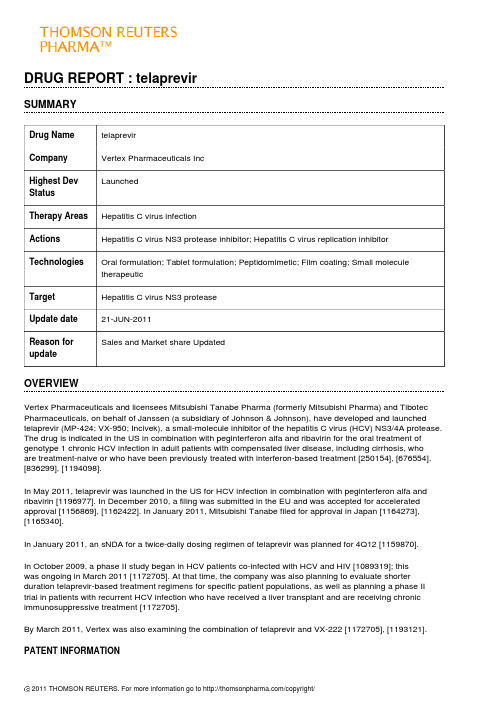
DRUG REPORT : telaprevirSUMMARYDrug Name telaprevirCompany Vertex Pharmaceuticals IncLaunchedHighest DevStatusTherapy Areas Hepatitis C virus infectionActions Hepatitis C virus NS3 protease inhibitor; Hepatitis C virus replication inhibitorTechnologies Oral formulation; Tablet formulation; Peptidomimetic; Film coating; Small moleculetherapeuticTarget Hepatitis C virus NS3 proteaseUpdate date21-JUN-2011Reason forSales and Market share UpdatedupdateOVERVIEWVertex Pharmaceuticals and licensees Mitsubishi Tanabe Pharma (formerly Mitsubishi Pharma) and Tibotec Pharmaceuticals, on behalf of Janssen (a subsidiary of Johnson & Johnson), have developed and launched telaprevir (MP-424; VX-950; Incivek), a small-molecule inhibitor of the hepatitis C virus (HCV) NS3/4A protease. The drug is indicated in the US in combination with peginterferon alfa and ribavirin for the oral treatment of genotype 1 chronic HCV infection in adult patients with compensated liver disease, including cirrhosis, whoare treatment-naive or who have been previously treated with interferon-based treatment [250154], [676554], [836299], [1194098].In May 2011, telaprevir was launched in the US for HCV infection in combination with peginterferon alfa and ribavirin [1196977]. In December 2010, a filing was submitted in the EU and was accepted for accelerated approval [1156869], [1162422]. In January 2011, Mitsubishi Tanabe filed for approval in Japan [1164273], [1165340].In January 2011, an sNDA for a twice-daily dosing regimen of telaprevir was planned for 4Q12 [1159870].In October 2009, a phase II study began in HCV patients co-infected with HCV and HIV [1089319]; thiswas ongoing in March 2011 [1172705]. At that time, the company was also planning to evaluate shorter duration telaprevir-based treatment regimens for specific patient populations, as well as planning a phase II trial in patients with recurrent HCV infection who have received a liver transplant and are receiving chronic immunosuppressive treatment [1172705].By March 2011, Vertex was also examining the combination of telaprevir and VX-222 [1172705], [1193121].PATENT INFORMATIONThe US patent covering the composition-of-matter for telaprevir was granted in 2010 with a term that expiresin 2025. In the EU, Vertex expected to obtain extensions to the term of the patent covering the composition-of-matter of telaprevir, thus extending expiry from 2021 to 2026; the company is required to apply separately for the extensions in the EU on a country-by-country basis [1172705].REGULATORY INFORMATIONThe USIn December 2005, the FDA granted telaprevir Fast Track status for HCV treatment [639367].By July 2010, a rolling NDA submission had been initiated; at that time, the CMC section of the NDA had been filed [1119626]. In November 2010, the filing was completed, which included data from both treatment-naive and -experienced HCV patients. The company also requested priority review [1149931], [1159870]. In January 2011, the NDA was accepted for priority review by the FDA, with a PDUFA date of May 23, 2011 [1162422].In April 2011, the FDA's Antiviral Drugs Advisory Committee recommended approval for genotype 1 chronic HCV infection [1187825]. In May 2011, telaprevir was approved for the treatment of chronic HCV infection in combination with peginterferon alfa and ribavirin in adult patients who had either not received interferon-based treatment or who had not responded adequately to prior therapy [1193767]. The drug was launched in the US later that month [1196977].In January 2011, an sNDA for a twice-daily dosing regimen of telaprevir was planned for 4Q12 [1159870]. EuropeIn December 2010, a filing was submitted in the EU for chronic HCV infection, which was accepted by the EMA for accelerated approval [1156869], [1162422]. In May 2011, a decision was anticipated in the second half of 2011 [1188291].JapanIn July 2010, filing was slated for early 2011 [1121439]. In January 2011, Mitsubishi Tanabe filed for approval in Japan [1164273].Rest of the worldIn January 2011, Vertex completed submission of its NDS for telaprevir in Canada; the drug was granted priority review in Canada [1162422].PREMARKETING STUDIESTreatment-naivePhase IIIIn March 2011, Vertex was planning a study to evaluate a short-duration, 12-week telaprevir-based regimen in treatment-naive patients with HCV with the CC IL28B genotype [1180816].In March 2011, a phase III HCV and HIV co-infection study of telaprevir was planned for the end of that year [1172677].In October 2010, the phase IIIb trial (OPTIMIZE) to compare twice-daily telaprevir (1125 mg) with three times-daily telaprevir (750 mg) was initiated in 700 treatment-naive patients with genotype-1 HCV in the US and the EU [1141995]. The randomized, open-label, study would have a primary endpoint of sustained viral response (SVR) 24 weeks after the end of all treatment. The primary objective would be to demonstrate non-inferiorityof twice-daily telaprevir versus telaprevir dosed every 8 h as measured by SVR. SVR data were expected in 2012. Patient screening for enrollment was expected to begin in November 2010 [1141996]. In January 2011, enrollment was ongoing [1159870].In October 2008, a randomized, open-label, active-control, parallel-group, safety and efficacy phase III trial (NCT00780416; G060-A6) was initiated in treatment-naive patients (expected n = 165) with genotype-1 HCV infection in Japan. Patients were to receive telaprevir in combination with peginterferon-alfa-2b (PEG-IFN-a-2b) plus ribavirin, or PEG-IFN-a-2b plus ribavirin alone. The primary endpoint was a sustained viral response(SVR) , defined as an undetectable HCV RNA level 24 weeks after treatment. The trial was expected to be completed in July 2011 [956738]. Development was ongoing in July 2009 [1047222].In September 2008, a randomized, open-label, active control, parallel group supplemental phase III trial(NCT00758043; VX08-950-111; ILLUMINATE) was initiated in treatment-naive patients (expected n = 500) with HCV at sites in the EU and US. Patients who achieved a rapid viral response to a telaprevir-based regimen were to be evaluated after extending total treatment duration from 24 to 48 weeks. The primary endpoint was SVR rate [956721], [1172705]. By January 2009, enrollment was complete [975836]. By August 2009, the telaprevir dosing section of the study had been completed and all patients were beyond week 24 [1031951]. In August 2010, data were reported, which demonstrated that there was no benefit from extending the telaprevir-based treatment to 48 weeks. The SVR rates were 92% (n = 149/162) and 88% (n = 140/160) for the 24 and 48-week treatment groups respectively. Overall, in the intent-to-treat (ITT) analysis, 72% of patients (n = 388/540) in the trial achieved a viral cure, and the safety profile was similar to the phase III ADVANCE study. The most common adverse events reported included fatigue, pruritus, nausea, anemia, rash and headache, and majority were considered mild or moderate [1123095]. In October 2010, data from the trial were presented at the 61st AASLD meeting in Boston, MA. It was demonstrated that 60% of African-Americans and 63% of all patientswith advanced liver fibrosis or cirrhosis achieved SVR with telaprevir-based therapy. Of African-Americans whose virus was undetectable at weeks 4 and 12, 88% achieved SVR in the 24- and 48-week randomized treatment arms. There was no control arm of PEG-IFN and ribavirin alone in the study. A total of 65% of people met the criteria for 24-week total treatment; there was no benefit in extending therapy to 48 weeks. Adverse events resulted in 17% of patients discontinuing the study, mainly due to a severe rash or anemia [1143419], [1146345].In March 2008, patient screening began in a randomized, double-blind, placebo-controlled, parallel-group, three-arm safety and efficacy phase III trial (NCT00627926; ADVANCE; VX07-950-108), which was expected to enroll 1050 treatment-naive patients with genotype-1 chronic HCV. The trial was to be conducted at 114 sites worldwide, including sites in the EU and US. The patients were to receive one of two regimens of telaprevir(8 weeks or 12 weeks) administered in combination with PEG-IFN-a-2a and ribavirin, or placebo. The primary endpoint was an SVR, defined as undetectable HCV RNA, 24 weeks after treatment completion [885649], [885933]. By October 2008, enrollment was completed [956858]. In February 2009, dosing of telaprevir or placebo, 8 or 12 weeks, had been completed [996403]. Data were reported in May 2010, showing that telaprevir was superior to control; SVR was achieved in 69 and 75% of telaprevir-treated patients after 8 and 12 weeks, respectively. After 48 weeks, 44% of patients in the control arm achieved SVR. Rapid viral responses were achieved by 68, 66 and 9% of patients in the 12-week telaprevir arm, the 8-week telaprevir arm and the control arm, respectively. Viral relapse was observed in 9, 10 and 28% of patients in the 12-week telaprevir arm, the8-week telaprevir arm and the control arm, respectively [1103157]. In October 2010, further data from the trial were presented at the 61st AASLD meeting in Boston, MA. It was demonstrated that 62% of African-Americans achieved SVR with telaprevir, compared to 25% of African-Americans treated with PEG-IFN and ribavirin alone.A total of 62% of people with advanced liver fibrosis or cirrhosis achieved SVR with telaprevir, compared to 33% treated with PEG-IFN and ribavirin alone. In the study, 58% of patients met the criteria for 24-week total treatment [1143419]. Further data presented at the meeting concluded that the 12-week regimen of teleprevir (150 mg/8 h) plus PEG-IFN (18 microg/week) and ribavirin (1000 to 1200 mg/day) provided the best benefitto risk profile. The most frequently observed adverse events were nausea, anemia, diarrhea, fatigue and rash [1148314].Phase IIIn May 2011, a US phase II trial to assess the efficacy of telaprevir in combination with PEG-IFN and ribavirin in patients with recurrent HCV following a liver transplant was planned for the fourth quarter of 2011 [1188291].In October 2009, a phase II multicenter, randomized, double-blind, placebo-controlled, two part (part A andpart B) parallel-group, safety and efficacy study (NCT00983853; VX08-950-110) began in the US in patients(n = 68) co-infected with HCV and HIV, who were treatment-naive for HCV. The study was to determine the efficacy of treatment with telaprevir plus peginterferon alfa-2a and ribavirin, with primary endpoints of safety and plasma HCV RNA level. The study was due to complete in June 2012 [1089319]. By October 2010, enrollment of 60 patients had been completed [1141995]. Patients in Part A and Part B of the study were randomized to receive either 12 weeks of telaprevir or placebo in combination with peginterferon alfa-2a and ribavirin followedby 36 weeks of peginterferon alfa-2a and ribavirin alone. Part A (n = 13) of the study enrolled patients who were not receiving antiretroviral therapy for HIV. Part B (n = 47) enrolled patients who were being treated for HIVwith either Atripla (n = 24) or a Reyataz-based regimen (n = 23). In March 2011, interim data (from 4 weeksof treatment) were presented at the 18th Conference on Retroviruses and Opportunistic Infections (CROI) in Boston, MA. The results demonstrated that 70% of patients in the study (Parts A and B) had undetectable HCV after treatment with telaprevir-based combination therapy compared to 5% in those on pegylated-interferonand ribavirin alone at week 4. In addition, HIV viral load and CD4 counts were stable in patients on a telaprevir-based regimen. Final data for sustained viral response (SVR) were expected in 2012 [1172677].In August 2009, a combination study of telaprevir and VX-222 was planned [1031951]. In October 2009, the trial was still expected to start that year, and data were expected by mid-2010 [1052205]. In January 2010, the trial was to begin pending completion of discussions with regulatory authorities [1067607]. In February 2010, the trial was planned to start in the first quarter of 2010 and interim data were expected in the third quarter of 2010. The trial would evaluate the rate of undetectable HCV RNA 24 weeks after the end of treatment with multiple VX-222 and telaprevir dosing regimens [1073316]. In March 2010, the phase II multicenter, randomized, parallel-group, dose-ranging trial (; VX09-222-103) began. The trial was designed to evaluate the safety and efficacy of telaprevir plus VX-222 in treatment-naive patients with genotype I HCV infection (expected n = 150), using a response-guided trial design. The primary endpoint of the trial was safety and tolerability, with a secondary endpoint of proportion of patients achieving SVR; the company stated that it may elect to enroll upto two additional treatment arms. At that time, initial data were expected in 2H10 [1078227], [1089256]. In April 2010, enrollment in the study was expected to be completed in the second quarter of 2010 and interim data were expected in the second half of 2010 [1091125]. In October 2010, Vertex discontinued arm A of the trial due to patients meeting a pre-defined stopping rule related to viral breakthrough in the first 4 weeks of dosing. At that time, an additional three cohorts were continuing and no viral breakthrough had been observed. Data were expected in the first half of 2011 with SVR data in the second half of 2011 [1141995]. In November 2010, the company planned to begin an additional treatment arm evaluating the triple combination regimen of telaprevir, VX-222 and ribavirin (po, bid) for 12-week using a response-guided regimen. Enrollment was expected to begin in the first quarter of 2011. The phase II study also included treatment arms evaluating 12-week, response-guided regimens of four-drug (telaprevir/VX-222 with or without Pegasys and ribavirin) combination therapy. Enrollment in the four drug treatment arm was completed in October 2010. Vertex may add an additional arm based on further results from the ongoing treatment arms [1146979]. In December 2010, the second two-drug treatment arm of telaprevir plus VX-222 (1125mg/400mg) alone was discontinued as a result of a pre-defined stopping rule related to viral breakthrough. The study would continue as planned with three treatment arms of the four-drug combinations, two of which were fully enrolled. At that time, enrollment in the three-drug treatment arm and data from the four-drug treatment arms were expected in the first quarter of 2011 [1157421]. In March 2011, interim data from the 106-patient ZENITH study were presented at the 46th annual meeting of the European Association for the Study of the Liver in Berlin, Germany. At week 12, 90 and 83% of patients treated with 400 and 100 mg VX-222, respectively, in combination with telaprevir, pegylated-interferon and ribavirin, showed undetectable hepatitis C virus. At that time, 50 and 38% of patients in the two groups, respectively, were able to stop all treatment. The remaining patients received a further 12 weeks of pegylated-interferon and ribavirin. At that time, enrollment was underway in an additional arm assessing VX-222, telaprevir and ribavirin [1180638].By February 2008, a phase II viral kinetics trial for HCV genotype-4 had begun [875818]. By July 2008, enrollment was complete [930471].In January 2008, an open-label, single-group, safety and efficacy phase II trial (NCT00621296) was initiatedin subjects (expected n = 16) with chronic HCV in Japan. The subjects were to receive telaprevir 750 mgthree times daily, for 24 weeks. The primary endpoint was to establish HCV RNA kinetics and other viral characteristics. The study was expected to be completed in September 2009 [907396], [904699]. Patient recruitment was ongoing in August 2008 [907396].In October 2007, a randomized, open-label, parallel-group, safety and efficacy phase II trial (NCT00528528;VX-950-TiDP24-C208; CR013516) was initiated in treatment-naive patients (expected n = 160) with genotype-1 chronic HCV in Europe. The patients were randomized to 1 of 4 treatment groups and received telaprevir (750 mg tid or 1125 mg bid) for 12 weeks in combination with PEG-IFN-a-2a plus ribavirin or PEG-IFN-a-2b plusribavirin for 24 weeks [956710]. In July 2008, data were released from the study. No substantial differences were seen in the safety profiles of the twice- and three-times-daily regimens, and both resulted in > 80% of patients with undetectable HCV RNA at weeks 4 and 12. A complete analysis was to be carried out when the study finished in 2009 [930471]. In October 2008, similar data were presented at the 59th Annual Meeting of the American Association for the Study of Liver Diseases in San Francisco, CA [958470]. In April 2009, all dosingin the study had been completed and in August 2009, patients were in the follow-up period. The number of patients with an SVR24 in the bid and tid dose groups would be compared [1031951]. In October 2009, further data were presented . An intent-to-treat analysis of 161 patients showed that SVR rates were 82 and 83% for patients receiving peg-IFN-alfa-2b bid and peg-IFN-alfa-2a bid, respectively, and 81 and 85% for those on peg-IFN-alfa-2b tid and peg-IFN-alfa-2a tid, respectively. RVR rates for the four groups were 67, 83, 69 and 80%, respectively. The most common adverse events were pruritis, rash, nausea, flu-like illness, anemia, headache and fatigue. These were similar in the bid and tid regimens. Of 161 patients, 5% discontinued treatment because of serious adverse events, namely rash and anemia. The data were to be presented at the 60th Annual Meeting of the American Association for the Study of Liver Diseases in Boston, MA [1053696], [1056258]. By January 2010, data from the trial had been submitted to the FDA and discussions were underway with bothUS and EU regulatory authorities regarding development of the drug as part of an every 12 h dosing regimen [1067607].In May 2006, Vertex was planning the phase II study PROVE 2 study in Europe. This would incorporate four,80-patient treatment arms: subjects were to receive telaprevir plus PEG-IFN for 12 weeks; telaprevir with PEG-IFN and ribavirin for 12 weeks; 12 weeks treatment with the triple combination followed by 12 weeks withPEG-IFN and ribavirin alone; or PEG-IFN and ribavirin for 48 weeks. The primary objectives were the sameas for PROVE 1 [668896]. By July 2006, PROVE 2 had started and by January 2007, recruitment had been completed [680324], [755789]. At that time, the company expected to expand clinical development of telaprevir into important HCV sub-populations in 2007, including patients with genotype-2 and genotype-3 HCV infection. The company also anticipated initiating clinical trials exploring twice-daily dosing of telaprevir some time in 2007 [755789]. In June 2007, preliminary data from the PROVE 2 study were reported to be consistent with PROVE 1 study results. Patients treated with telaprevir, PEG-IFN and ribavirin had undetectable HCV RNA levels at 4 and 12 weeks. At 12 weeks, treatment with telaprevir only was associated with antiviral activity that was lower than that in patients that received telaprevir with ribavirin and PEG-IFN, but was higher than that observed in the control arm. Rash, gastrointestinal events and anemia were the most common adverse events that led to discontinuation in the telaprevir arms. Fewer discontinuations were observed in patients that received telaprevir and PEG-IFN [803933]. In November 2007, similar data were presented at the 58th Annual Meeting of the American Association of the Study of Liver Diseases in Boston, MA. The SVR rates were 29, 59 and 65% in the 12-week (without ribavirin), 12-week (plus ribavirin) and 24-week treatment arms, respectively [847055]. In April 2008, similar data from the trial were presented at the 43rd Annual Meeting of the European Association for the Study of the Liver in Milan, Italy [899125]. Similar data were also reported in May 2008 at the DDW meetingin San Diego, CA [906212]. In October 2008, similar data were presented at the 59th Annual Meeting of the American Association for the Study of Liver Diseases in San Francisco, CA. The SVR rates were 30, 60 and69 and 46% in the 12-week (without ribavirin), 12-week (plus ribavirin), 24-week, and 48-week treatment arms, respectively [958470]. In April 2009, similar data were published [1004842], [1004581].In October 2005, Vertex was planning one- and three-month phase II studies of telaprevir in combination with PEG-IFN [626769]. By November 2005, Vertex had submitted an IND to the FDA [634055]. In December 2005, Vertex began a 28-day, US phase II study to evaluate the safety, tolerability and pharmacodynamics of 750 mg of telaprevir given every 8 h in combination with PEG-IFN and ribavirin in 12 treatment-naive subjects [638638]. In January 2006, enrollment was complete [644067]. In February 2006, Vertex reported preliminary results showing that at the end of week 1, plasma HCV RNA was below the limit of quantitation in half of the subjects and undetectable in 2 of the patients. After 4 weeks of dosing, HCV RNA was undetectable in plasma from all 12 patients. There were no treatment discontinuations and no serious adverse events reported [648823]. In May 2006, similar data were presented at the Discovery & Selection of Successful Drug Candidates meeting in Cambridge, MA [667947]. Also in May 2006, similar data were presented at the DDW meeting in Los Angeles, CA. All patients reached undetectable plasma HCV RNA levels within 28 days of dosing and levels remained undetectable after 12 weeks follow-up [667643], [668532].Treatment-experiencedPhase IIIIn October 2008, an open-label, uncontrolled, single-group, safety and efficacy phase III trial (NCT00781274;G060-A9) was initiated in patients (expected n = 30) with genotype-1 HCV who did not respond to previous therapy in Japan. Patients were to receive telaprevir for 12 weeks and peginterferon alfa-2b (PEG-IFN-a-2b) plus ribavirin for 24 weeks. The primary endpoint was an SVR, defined as an undetectable HCV RNA level 24 weeks after treatment. The trial was expected to be completed in January 2011 [956789].In October 2008, an open-label, uncontrolled, single-group, safety and efficacy phase III trial (NCT00780910;G060-A8) was initiated in patients (expected n = 100) with genotype-1 HCV who relapsed following previous therapy in Japan. Patients were to receive telaprevir for 12 weeks and PEG-IFN-a-2b plus ribavirin for 24 weeks. The primary endpoint was a SVR, defined as an undetectable HCV RNA level 24 weeks after treatment. The trial was expected to be completed in January 2011 [956781].In August 2008, Vertex reached agreement with US and EU regulatory authorities to proceed with its planned REALIZE trial (NCT00703118; CR014842) [936309], [956718]. In the 48-week, 650-patient, three-armed multicenter study patients with genotype-1 HCV who had failed to achieve a SVR with prior therapy of PEG-IFN plus ribavirin, would receive 750 mg of telaprevir q8 h plus PEG-IFN-a-2a and ribavirin for 12 weeks, followed by 36 weeks of PEG-IFN-a-2a plus ribavirin alone. Patients in the control arm would receive PEG-IFN-a-2a plus ribavirin alone for 48 weeks. The primary endpoint was undetectable HCV RNA levels 24 weeks after the endof treatment [936309]. By October 2008, patient dosing had begun; the trial was to include null responders, partial responders and relapsers to prior therapy [952629], [956585], and in November 2008, enrollment in Europe began [965253]. In February 2009, enrollment in REALIZE was complete [982641]. By August 2009,the telaprevir dosing section of the study had been completed and all patients were beyond week 24 [1031951]. In September 2010, top-line data were reported, which demonstrated that 65% of HCV patients (total n = 622) treated with telaprevir achieved a SVR compared to 17% in the control group at 24 weeks. In a subgroup analysis, data showed that 86% of relapsed patients treated with telaprevir achieved a SVR compared to 24% for the control. In addition, the SVR rates were 57% and 31% for partial and null responders, respectively, compared to 15% and 5% for the corresponding control arms [1143647]. In March 2011, further data were presented at the International Liver Congress 2011/46th annual meeting of the European Association for the Study of the Liver (EASL) in Berlin, Germany. No significant improvement in SVR rates and no significant reduction in virologic failure and relapse rates were observed in patients treated with a 4-week lead-in of PEG-IFN-a-2a and ribavirin alone compared with a simultaneous start of telaprevir, PEG-IFN-a-2a and ribavirin [1180788], [1184383].In February 2007, phase III trials were planned, and development of the drug was to be expanded to include HCV genotype-2 and -3 and twice-daily dosing schedules [762718], [762718]. In April 2007, Vertex and Tibotec planned to meet with regulatory authorities to discuss the design of phase III trials [784124], and by October 2007, Vertex and Tibotec were in discussions with the US and EU regulatory authorities to review clinical data and to ascertain how best to evaluate the drug in future trials [845344]. By January 2008, Vertex had submitted a phase III protocol to the FDA. The design included a 48-week control arm and 8 and 12 weeks of telaprevir treatment as part of 24-week combination treatment regimens. Vertex planned to determine which patients should cease treatment at week 24 using rapid viral response (RVR). A meeting with the FDA was scheduled for January 2008 to discuss the protocol [865541].In May 2006, the company began PROVE 1, a randomized, US phase II study, which was expected to enroll 280 patients into 4 treatment arms: 20 patients would receive 750 mg of telaprevir three times a day with PEG-IFN and ribavirin for 12 weeks; two 80-patient groups would take the same combination for 12 weeks followed by 12 or 36 weeks of treatment with PEG-IFN and ribavirin alone; the fourth group of 80 patients would receive PEG-IFN and ribavirin alone for 48 weeks. The primary objective was to determine the number of patients who achieved SVR, defined as undetectable HCV RNA levels 24 weeks after completing treatment. Patients in the 12- and 24-week treatment arms who had undetectable viral levels by the end of week 4 and who maintained this until week 10 or 20, respectively, would stop treatment at 12 or 24 weeks, respectively. Patients who had detectable HCV RNA at the initial time point would continue on PEG-IFN and ribavirin for 48 weeks. Enrollment began in June 2006 [668896]; by September 2006, enrollment in the trial had been completed [735053]. In December 2006, Vertex reported interim data from the study. An independent board compared pooled data from the three telaprevir arms with the control arm. At 12 weeks, 88% telaprevir-treated patients had undetectable HCV RNA levels, compared with 52% patients in the control group. In the telaprevir arms, the discontinuationrate was 9%, compared with 3% in the control arm. Serious adverse events occurred in 3% of telaprevir-treated patients and 1% of patients in the control group [751574], [751209]. In April 2007, further data were presented at the 42nd annual meeting of the European Association for the Study of the Liver in Barcelona, Spain, showing 88 and 79% of patients that received telaprevir achieved plasma HCV RNA levels of < 30 IU/ml and < 10 IU/ ml, respectively, at 4 weeks. Furthermore, 6 of 9 patients in one treatment arm continued to have undetectable HCV RNA 20 weeks after stopping treatment [784124]. In September 2007, data were presented at the 47th ICAAC meeting in Chicago, IL. Interim, 24-week PROVE 1 data demonstrated that telaprevir patients (n = 175) achieved undetectable HCV RNA at weeks 4 (79%) and 12 (70%); in the control arm (n = 75) 11% achieved undetectable HCV RNA at weeks 4 and 39% at week 12. Discontinuations due to adverse events were more frequent in the telaprevir arm at 11%, versus 35% in the control arm [820890]. In November 2007, similar data were presented at the 58th Annual Meeting of the American Association of the Study of Liver Diseases in Boston, MA [847055]. In April 2008, final data from the trial were presented at the 43rd Annual Meeting of the European Association for the Study of the Liver in Milan, Italy. SVRs were seen in 61% of telaprevir recipients, compared with 41% of control patients [899125]. Similar data were reported in May 2008 at the DDW meeting in San Diego, CA [907429]. In April 2009, similar data were published [1004828], [1004581].Phase IIIn September 2007, a non-randomized, open label, uncontrolled, single-group, phase II study (NCT00535847; study 107; VX06-950-107) was initiated in the US, Canada, Germany, France, the Netherlands and Puerto Rico. In the trial, 170 patients with genotype-1 hepatitis C who did not achieve an SVR with previous interferon-based treatment in the control arms of the PROVE 1, PROVE 2 or PROVE 3 trials were to receive telaprevir plus PEG-IFN-a-2a plus ribavirin for 12 weeks, followed by the same regimen without telaprevir for 12 weeks. The trial was expected to be completed in May 2009 [898809], [898686]. Interim data were reported in April 2008 at the 43rd Annual Meeting of the European Association for the Study of the Liver in Milan, Italy. Of the 60 patients treated with the telaprevir combination, 49 achieved HCV RNA levels < 25 IU/ml. This response was maintained, and no viral breakthrough was observed in 36 and 16 patients who had completed 4 and 12 weeks of treatment, respectively. Two patients discontinued treatment due to adverse events, which were similar to those commonly observed with PEG-IFN-a-2a and ribavirin. They included fatigue, nausea, rash and headache [899404]. In October 2008, further data were presented at the 59th Annual Meeting of the American Association for the Study of Liver Diseases in San Francisco, CA. At 4, 12 and 24 weeks, 65.4, 65.6 and 56.9% patients had HCV RNA <10 IU/ml [958470]. In October 2009, further interim data from 94 patients were reported. An SVR was achieved after 24 or 48 weeks of telaprevir-based treatment, by 90% of prior treatment responders and 55% of prior treatment partial responders. After 48 weeks of telaprevir-based treatment, an SVR was achieved by 57% of prior treatment null responders. At that time, adverse events had caused 7% patientsto discontinue treatment [1052655]. In April 2010, data were presented at the 45th Annual Meeting of the European Association for the Study of the Liver in Vienna, Austria. An overall SVR rate of 59% and an overall relapse rate of 16% were observed. In addition, it was reported that 10 patients had discontinued all therapy due to adverse events, with rash being the most common adverse event [1091068]. Further data were presented in May 2010 at the DDW meeting in New Orleans, LA. The viral breakthrough and relapse rates were 25 and 23% in prior null responders, 10 and 22% in prior partial responders, 13 and 0% in prior viral breakthroughs, and 0 and 4% in prior relapsers, respectively [1091233].By January 2007, Tibotec had begun a European phase II viral kinetics trial. The study would evaluate telaprevir in patients with HCV genotypes 2 and 3. At that time, patients were undergoing screening [865541]. By July 2008, enrollment was complete [930471].By October 2006, Vertex was planning PROVE 3, a 400-patient trial in patients with genotype-1 HCV who had failed prior treatment [735053]. In February 2007, Vertex began the phase IIb PROVE 3 trial. The randomized, multicenter, 440-patient, North American and European study assessed 750 mg of telaprevir every 8 h in patients with interferon-refractory, genotype-1 HCV. Patients received one of four treatment regimes: 12 weeks of telaprevir, PEG-IFN alfa-2a and ribavirin, followed by 12 weeks of PEG-IFN-a-2a and ribavirin; 24 weeksof telaprevir and PEG-INF-a-2a; 24 weeks of telaprevir, PEG-IFN-a-2a and ribavirin, followed by 24 weeks of interferon PEG-IFN-a-2a and ribavirin; or 48 weeks of PEG-IFN-a-2a and ribavirin. Patients in the final arm had the option to receive telaprevir if they did not respond to PEG-IFN-a-2a plus ribavirin [762718]. Enrollment of 440 patients was completed in June 2007 [803933]. In January 2008, Vertex was planning to meet with the FDA to discuss the telaprevir development program, once interim results from PROVE 3 were available [865541]. In June 2008, interim data were reported. By that time, patient dosing had been completed. In the first treatment。

药物Dasabuvir(达萨布韦)合成检索总结报告
一、Dasabuvir(达萨布韦)简介
Dasabuvir(达萨布韦)于2014年12月19日在美国上市。
Dasabuvir (达萨布韦)是HCV NS5B RNA-依赖RNA聚合酶抑制剂,适用于基因1型慢性丙肝感染。
Dasabuvir(达萨布韦)不良反应有:疲劳、瘙痒、感觉虚弱或缺乏能量、恶心及失眠。
Dasabuvir(达萨布韦)分子结构式如下:
英文名称:Dasabuvir 英文名称:Dasabuvir sodium salt
中文名称:达萨布韦中文名称:达萨布韦钠盐本文主要对Dasabuvir(达萨布韦)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Dasabuvir(达萨布韦)合成路线
三、Dasabuvir(达萨布韦)合成检索总结报告(一) Dasabuvir(达萨布韦)中间体2的合成
(二) Dasabuvir(达萨布韦)中间体3的合成
(三) Dasabuvir(达萨布韦)中间体5的合成方法一。

药物Velpatasvir(维帕他韦)合成检索总结报告一、Velpatasvir(维帕他韦)简介Velpatasvir(维帕他韦)于2016年6月在美国上市,主要用于治疗成人慢性HCV基因型1、2、3、4、5或6的感染。
Velpatasvir(维帕他韦)常见的不良反应有头疼、疲劳、贫血、恶心、失眠和腹泻。
Velpatasvir(维帕他韦)分子结构式如下:CAS:1335316-40-9英文名称:Velpatasvir中文名称:维帕他韦本文主要对Velpatasvir(维帕他韦)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Velpatasvir(维帕他韦)合成路线三、Velpatasvir (维帕他韦)合成检索总结报告(一)Velpatasvir (维帕他韦)中间体3的合成方法一合成方法实验步骤参考文献操作方法一Take 4-bromo-3-bromomethylacetophenone (2)290g,7-hydroxy-tetralone (1)210g,potassium carbonate 350g,acetonitrile 700ml,Benzene 700ml,react at room temperature overnight,continue to react at 60°C for 3h,filter at the end of the reaction,The filtrate was decompressed to recover the solvent.The residue was added with water (500mL)and extracted with ethyl acetate (500mL).The organic phase was washed with saturated brine and dried over anhydrous sodium sulfate.The crude product was concentrated and added with petroleum ether.The mixture was filtered with suction and the filter cake was dried to obtain 339g of a yellow solid 3with a yield of 91%.CN107629029;(2018);(A)Chinese 操作方法二A mixture of 1-(4-bromo-3-(bromomethyl)phenyl)ethanone 2(500mg,1.71mmol),7-hydroxytetralone 1(291mg,1.79mmol),potassium carbonate (472mg,3.42mmol)and acetonitrile (5mL)was heated at about 70°C.for about 2h.The mixture was partitioned between MTBE (20mL)and water (2×10mL).The organic layer was dried over anhydrous sodium sulfate,filtered and concentrated under reduced pressure to give crude product.Column chromatography of the crude mixture on silica gel using 15-50%ethyl acetate hexanes gradient followed by recrystallization from methanol (3.5mL)gave Compound (3).US2015/361073;(2015);(A1)English 操作方法A compound of formula 2(18g),potassium carbonate (25.32g),and tetrabutylammonium bromide (300mg)were added to a solution of 7-hydroxyltetralone 1(10g)in toluene (90mL)and dimethylsulfoxide (10mL).The reaction mixture was stirred at 40°C for 2hours.After completion ofWO2017/60820;(2017);(A1)三the reaction,the reaction mixture was filteredand washedwith carbon tetrachloride.Water (100mL)was added to the filtrate and stirred for 10minutes.The organic layer was separated and concentrated to afford a compound of formula 3as an off-white solid.English(二)Velpatasvir (维帕他韦)中间体3的合成方法二合成方法实验步骤参考文献操作方法一A 100mL three-necked flask was charged with a compound 7-hydroxy-3,4-dihydro-2H-1-naphthalenone 1(37.68g,232.32mmol)and 38ml of N,N-dimethylformamide,Potassium carbonate (41.88g,303.03mmol)and potassium iodide (3.35g,20.18mmol)were added and the mixture was stirred for 30minutes.The compound of formula 4(50g,202.02mmol)was further added and reacted at 20-25o C for 18h.After the completion of the TLC control reaction,76ml of water was quickly added dropwise to the reaction solution to control the temperature at a temperature of ≦25°C,a large amount of solid was precipitated,stirred for 1hour,filtered,rinsed with water and dried at 55-60°C to obtain 74.35g of of compound 3(off-white solid),yield:98.6%.CN107501229;(2017);(A)Chinese 操作方法二A mixture of 1-(4-bromo-3-(chloromethyl)phenyl)ethanone 4(600mg,2.42mmol),7-hydroxytetralone 1(393mg,2.42mmol),potassium carbonate (668mg,4.84mmol)and tetrabutylammonium bromide (78mg,0.242mmol)in DMAc (3mL)was stirred at room temperature for about 20h.The reaction mixture was partitioned between ethyl acetate (18mL)and water (2×6mL).The organic layer was dried over anhydrous sodium sulfate and concentrated to an approximate volume of 5mL.Product precipitate was filtered,washed with ethyl acetate (2mL)and dried under vacuum to give Compound 2015/361073;(2015);(A1)English(三)Velpatasvir (维帕他韦)中间体5的合成。

药物Doravirine(多拉维林)合成检索总结报告一、Doravirine(多拉维林)简介Doravirine(多拉维林)是由时默沙东公司研发,于2018年8月在美国上市,主要与其他抗逆转录病毒药物联合用于治疗无抗逆转录病毒治疗史的成人患者的HIV-1感染。
Doravirine(多拉维林)是通过非竞争性的抑制HIV-l逆转录酶(RT)来抑制HIV-1复制。
Doravirine(多拉维林)不良反应:恶心,头晕,头痛,疲劳,腹泻,腹痛和做异常梦。
Doravirine(多拉维林)分子结构式如下:CAS:1338225-97-0英文名称:Doravirine中文名称:多拉维林二、Doravirine(多拉维林)合成路线三、Doravirine (多拉维林)合成检索总结报告(一)Doravirine (多拉维林)中间体3的合成合成方法实验步骤参考文献合成方法一To a round bottom flask charged with 3-bromo-5-chloro-phenol 2(5.20g,25.1mmol)and potassium carbonate (3.46g,25.1mmol)was added N-methylpyrrolidinone (25mL).To this suspension under N 2was added 2-chloro-3-fluoro-4-(trifluoromethyl)pyridine 1(5.00g,25.1mmol)and the reaction mixture was placed in an oil bath at 120o C.After 60minutes,the reaction mixture was allowed to cool to room temperature at which point,water (100mL)was added.The mixture was extracted with ethyl acetate (2x 100mL)and the combined organic fractions were washed with brine (3x 100mL),dried (MgSO 4),filtered and the solvent was evaporated under reduced pressure.The resulting residue was adsorbed onto silica gel and purified by column chromatography on a pre-packed silica gel Redi Sep 330gram column,eluting with 0-75%CH 2Cl 2in hexanes to yield the title compound 3.WO2009/67166;(2009);(A2)English A mixture of the 3-bromo-5-chlorophenol 2(3.74g;18.0合成方法二mmol),2-chloro-3-fluoro-4-(trifluoromethyl)pyridine1(3.00g;15.0mmol)and K2CO3(2.49g;18.0mmol)in NMP(15mL)was heated to120°C.for one hour,then cooled to room temperature.The mixture was then diluted with250mLEtOAc and washed with3×250mL1:1H2O:brine.Theorganic extracts were dried(Na2SO4)and concentrated invacuo.Purification by ISCO CombiFlash(120g column;load with toluene;100:0to0:100hexanes:CH2Cl2over40minutes)provided title compound3as a white solid.Repurification of the mixed fractions provided additionaltitle compound.US2011/245296;(2011);(A1)English(二)Doravirine(多拉维林)中间体4的合成合成方法实验步骤参考文献合成方法一To a suspension of3-(3-bromo-5-chlorophenoxy)-2-chloro-4-(trifluoromethyl)pyridine3(3.48g;8.99mmol)int-BuOH(36mL)was added KOH(1.51g;27.0mmol)andthe mixture was heated to75°C.overnight,at which point ayellow oily solid had precipitated from solution,and LCMSanalysis indicated complete conversion.The mixture wascooled to room temperature,and neutralized by the additionof50mL saturated aqueous NH4Cl.The mixture was dilutedwith50mL H2O,then extracted with2×100mL EtOAc.Thecombined organic extracts were dried(Na2SO4)andconcentrated in vacuo.Purification by ISCO CombiFlash(120g column;dry load;100:0to90:10CH2Cl2:MeOH over40minutes)provided the title com‐pound4as a fluffy white solid.US2011/245296;(2011);(A1)English合成方法二To a round bottom flask charged with3-(3-bromo-5-chlorophenoxy)-2-chloro-4-(trifluoromethyl)pyridine3(9.1g,25.3mmol)and potassium hydroxide(3.96g,70.5mmol)was added tert-butanol(100mL).This suspension wasplaced in an oil bath at75o C.After48hours,the reactionmixture was allowed to cool to room temperature and wasquenched with saturated aqueous ammonium chloride(50mL)and diluted with water(50mL).The mixture wasextracted with ethyl acetate(2x100mL)and the combinedWO2009/67166;(2009);(A2)Englishorganic fractions were washed with water (3x 100mL).The solvent was evaporated under reduced pressure to yield a solid.This was adsorbed onto silica and purified by column chromatography on a prepacked silica gel Redi Sep 330gram column,eluting with 0-5%methanol in CH 2Cl 2to give the title compound 4.(三)Doravirine (多拉维林)中间体5的合成方法一合成方法实验步骤参考文献合成方法一A suspension of 3-(3-bromo-5-chlorophenoxy)-4-(trifluoromethyl)pyridin-2(lH)-one 4(10g,27.1mmol),zinc (0.089g,1.357mmol),palladium acetate (0.305g,1.357mmol)and l,l'-bis(diphenylphosphino)ferrocene (0.827g,1.492mmol)in DMA (560mL)was degassed,placed under N2,and then charged with zinc cyanide (1.593g,13.57mmol).This mixture was placed in an oil bath maintained at 90°C for 16hours,after which the reaction mixture was allowed to cool to room temperature.Water (200mL)was added and the mixture was filtered,washing the solids with water.The solids were allowed to air dry and were crystallized from acetomtrile (320mL)to give Compound 5.WO2011/126969;(2011);(A1)English 合成方法二To a round bottom flask charged with 3-(3-bromo-5-chloro-phenoxy)-4-(trifluoromethyl)pyridin-2(lH)-one 4(3.00g,8.14mmol)and copper (I)cyanide (7.29g,81.0mmol)was added N-methylpyrrolidinone (25mL).This suspension under N 2was placed in an oil bath at 175o C.After 5hours the reaction mixture was allowed to cool to room temperature.Glacial acetic acid (30mL)was added to the mixture and stirred for 10minutes.The mixture was diluted with ethyl acetate (100mL),filtered through diatomaceous earth and the pad was washed with ethyl acetate (100mL).The filtrate was washed with water (3x 100mL)and brine (2x 100mL).The organic fraction was dried (Na 2SO 4),filtered and the solvent was removed under reduced pressure to yield a solid.This was adsorbed onto silica gel and purified by column chromatography on a pre-packed silicaWO2009/67166;(2009);(A2)Englishgel Redi Sep 120gram column,eluting with 0-5%methanol in CH2Cl 2to yield the title compound 5.(四)Doravirine (多拉维林)中间体5的合成方法二(1)Doravirine (多拉维林)中间体8的合成合成方法实验步骤参考文献合成方法一Ester 7(25.01g,104.4mmol,1.00equiv)was charged to toluene (113.43g,131mL,5.24vol)and 4-ethoxy-l,l,l-trifluoro-3-buten-2-one 6(26.43g,157.2mmol,1.51equiv)was added.The flow reactor consisted of two feed solution inlets and an outlet to a receiving vessel.The ester solution was pumped to one flow reactor inlet.Potassium tert-pentoxide solution was pumped to the second reactor inlet.Trifluoroacetic anhydride was added continuously to the receiver vessel.Triethylamine was added continuously to the receiver vessel.The flow rates were:13mL/min ester solution,7.8mL/min potassium tert-pentoxide solution,3.3mL/min trifluoroacetic anhydride and 4.35mL/min triethylamine.Charged toluene (50mL,2vol)and potassium trifluoroacetate (0.64g,4.21mmol,0.04equiv)to the receiver vessel.The flow reactor was submerged in a -10°C bath and the pumps were turned on.The batch temperature in the receiver vessel was maintained at 5to 10°C throughout the run using a dry ice/acetone bath.After 13.5min the ester solution was consumed,the reactor was flushed with toluene (10mL)and the pumps were turned off.The resulting yellow slurry was warmed to room temperature and aged for 4.5h.Charged methanol (160mL)to afford a homogeneous solution which contained 81.20area percent diene 8by HPLC analysis.The solution of diene 8(573mL)was used without purification in the subsequent reaction.WO2014/89140;(2014);(A1)English (2)Doravirine (多拉维林)中间体5的合成(由8合成5)合成方法实验步骤参考文献合成方法一To a solution of diene 8in PhMe/MeOH(573mL;40.69g,104.4mmol)was charged methanol (25mL,0.61vol).Ammonia (32g,1.88mol,18equiv)was added and the solution was warmed to 60°C.The reaction was aged at 60°C for 18h.The temperature was adjusted to 35-45°C and the pressure was decreased maintain a productive distillation rate.The batch volume was reduced to -300mL and methanol (325mL,8vol)was charged in portions to maintain a batch volume between 250and 350mL.The heating was stopped and the system vented.The resulting slurry was cooled to room temperature and aged overnight.The batch was filtered and the cake washed with methanol (3x,45mL).The wet cake was dried on the frit with suction under a nitrogen stream to afford 18.54g of a white solid 5.WO2014/89140;(2014);(A1)English;WO2015/84763;(2015);(A2)English(五)Doravirine (多拉维林)中间体5的合成方法三(1)Doravirine (多拉维林)中间体10的合成合成方法实验步骤参考文献合成方法一reaction mixture of 2-chloro-3-fluoro-4-(trifluoromethyl)-pyridine 1(11,5g,25mmol)and 3-chloro-5-hydroxy-benzonitrile 9(4.62g,30mmol)in 50mL of N-methyl pyrrolidinone (NMP)in the presence ofpotassium carbonate (4.16g,30mmol)was stirred at 80°C for 8h.The solution was cooled to room temperature,and 100mL of ice water wasadded to it slowly.Then the solid was filtered,washed with 20mL of DMF/water (1:1),and dried to give 3-chloro-5-((2-chloro-4-(trifluoromethyl)pyridin-3-yl)oxy)Bioorganic and Medicinal Chemistry ;vol.27;nb.3;(2019);p.447-456benzonitrile (10).Yield:90.5%,mp:163-165°C(2)Doravirine (多拉维林)中间体5的合成(由10合成5)合成方法实验步骤参考文献合成方法一Intermediate 10(1g,3mmol)and sodium hydroxide (0.60g,15mmol)were added to 20mL of methylbenzene and reacted at 150°C for 105min by the microwave method.The solvent was removed underreduced pressure,and then water (50mL)was added,neutralized to pH=7with hydrochloric acid.Then the residue was filtered and washed with water and ethyl acetate,and recrystallized from ethylacetate/normal hexane to afford the intermediate 3-chloro-5-((2-oxo-4-(trifluoromethyl)-1,2-dihydropyridin -3-yl)oxy)benzonitrile (5).Yield:54.1%,mp:273-274°C.Bioorganic and Medicinal Chemistry ;vol.27;nb.3;(2019);p.447–456.(六)Doravirine (多拉维林)中间体12的合成合成方法实验步骤参考文献合成方法一A suspension of the 3-chloro-5-{[2-hydroxy-4-(trifluoro-methyl)pyridin-3-yl]oxy}benzonitrile 5(2.00g;6.36mmol),5-(chloromethyl)-2,4-dihydro-3H-1,2,4-triazol-3-one 11(0.849g;6.36mmol)and K 2CO 3(0.878g;6.36mmol)in DMF (32mL)was stirred for 2hours at room temperature,at which point LCMS analysis indicated complete conversion.The mixture was diluted with 200mL Me-THF and washed with 150mL 1:1:1H 2O:brine:saturated aqueous NH 4Cl,then further washed with 2×150mL 1:1H 2O:brine.The aqueous fractions were further extracted with 150mL Me-THF,then the combined organic extracts were dried (Na 2SO 4)and concentrated in vacuo.Purification by ISCO CombiFlash (80US2011/245296;(2011);(A1)Englishg column;dry load;100:0to 90:10EtOAc:EtOH over 25minutes)provided the title compound 12as a white solid.合成方法二Pyridone 5(121kg,386.5mol)is dissolved in DMF (475L)and cooled between −10−10°C.To this solution is added potassium carbonate (106.9kg,773.5mol)followed by 5-chloromethyl-2,4-dihydro-[1,2,4]triazol-3-one 11(54.4kg,407.4mol)as a solution DMF (235L)over 7−9h.After aging for a further 0.5−1h,HPLC analysis revealed >97%conversion.4N HCl is then added (230L)slowly while keeping the temperature between 10and 30°C,followed by 600L of water.pH analysis revealed the pH to be ∼8.The mixture was aged for another 2h at this pH which was then further reduced to between 5and 6by addition of more 4N HCl (100L)and water (615L).The resulting slurry wasaged at between 10and 30°C for 10h,and then the solids were fifiltered.The wet cake is fifiltered and washed with 270L of water.The solids were then dried under vacuum at 65−75°C for 12h resulting in 193.7kg of crude product.The crude solid was then redissolved in a 19:1mixture of MeCN−H 2O (3000L)by heating the mixture to 80−90°C.The solution is then concentrated to 330−400L,and another 2500L of MeCN is added.Distillation is continued back down to a volume of 330−400L at which point the slurry is cooled and aged for 10h at a temperature between 10and 30°C.The slurry is then fifiltered and the wet cake washed with 100L of MeCN.The solids were dried under vacuum at 65−75°C for 12h,affffording the desired penultimate triazolone intermediate anic Process Research and Development ;vol.20;nb.8;(2016);p.1476-1481(七)Doravirine (多拉维林)13的合成方法一合成方法实验步骤参考文献The triazolone intermediate 12(130.6kg,317.2mol),NMP合成方法一(850L),potassium carbonate(65.9kg,477.5mol),andwater(5L)were charged to the vessel and degassed with N2.The mixture was cooled to5−10°C,and methyl iodide(50.9kg,381.2mol)is added over0.5h.The mixture is then agedfor34h after which point the reaction had reached∼90%conversion.The mixture was fifiltered over15kg of Celiteand the cake washed with375L of NMP.The resultingfiltrate was added over2.5h to water(2400L)at20−25°C,and the slurry was aged for24−30h at20−30°C.The slurryis then filtered and the wet cake washed with2000L ofwater.The wet cake is then taken up in450L of EtOH andheated to70−80°C for1.5−2h.After cooling to5−10°C thesolid was fifiltered,rinsed with EtOH,and dried undervacuum to afford105.4kg of crude doravirine13.The crudesolid is then recrystallized by slowaddition of EtOH(45L)to a80−95°C solution of1in NMP(320L),then seeding with crystalline1(121g)and furtheraddition of EtOH(13L).The mixture was cooled to between70and80°C and aged for2h.Then1400L of EtOH isadded.After aging for1h,the mixture is cooled to between5and10°C for7h.The slurry is then fifiltered and rinsedwith cold EtOH(45L).The solids were dried in a vacuumunder reduced pressure between50and75°C for17h,affording pure doravirine13.Organic ProcessResearch andDevelopment;vol.20;nb.8;(2016);p.1476–1481.合成方法二The triazolone intermediate12(2.37g;5.76mmol)andK2CO3(0.796g;5.76mmol)in DMF(58mL)was cooled to0°C.,then methyl iodide(0.360mL;5.76mmol)was added.The mixture was allowed to warm to room temperature,andstirred for90minutes,at which point LCMS analysisindicated>95%conversion,and the desired product of75%LCAP purity,with the remainder being unreacted startingmaterial and bis-methylation products.The mixture wasdiluted with200mL Me-THF,and washed with3×200mL1:1H2O:brine.The aqueous fractions were further extractedwith200mL Me-THF,then the combined organic extractswere dried(Na2SO4)and concentrated in vacuo.Theresulting white solid was first triturated with100mL EtOAc,then with50mL THF,which provided(after drying)the titlecompound13of>95%LCAP.Purification to>99%US2011/245296;(2011);(A1)English(八)Doravirine(多拉维林)13的合成方法二合成方法实验步骤参考文献合成方法一To a75L flask was charged a9.63wt%solution of3-(chloromethyl)-4-methyl-lH-l,2,4-triazol-5(4H)-one14inNMP(11.6kg,7.55mol),3-chloro-5-((2-oxo-4-(trifluoromethyl)-1,2-dihydropyridin-3-yl)oxy)benzonitrile5(2.00kg,6.29mol),NMP(3.8L)and2-methyl-2-butanol(6.0L).To the resulting suspension wasslowly added N,N-diisopropylethylamine(4.38L,25.2mol)over4h.The reaction was aged18h at ambient temperature.The reaction is considered complete when HPLC indicates<1%3-chloro-5-((2-oxo-4-(trifluoromethyl)-l,2-dihydro-pyridin-3-yl)oxy)benzonitrile remaining.The tan solutionwas quenched with acetic acid(1.26L,22.0mol)and aged atambient temperature overnight.The tan solution waswarmed to70°C.Water(2.52L)was added and the batchwas seed with anhydrate Form II(134g).The thinsuspension was aged lh at70°C.Additional water(14.3L)was added evenly over7h.The slurry was aged2h at70°Cand then slowly cooled to20°C over5h.The slurry wasfiltered and washed with2:1NMP/water(6L),followed bywater washes(6L x2).The filter cake was dried over a2sweep to give2.53kg(85%yield-corrected)of a whitesolid13that was confirmed to be crystalline Form II byX-ray powder detraction analysis.WO2014/89140;(2014);(A1)English;WO2015/84763;(2015);(A2)English。

药物Doravirine(多拉维林)合成检索总结报告一、Doravirine(多拉维林)简介Doravirine(多拉维林)是由时默沙东公司研发,于2018年8月在美国上市,主要与其他抗逆转录病毒药物联合用于治疗无抗逆转录病毒治疗史的成人患者的HIV-1感染。
Doravirine(多拉维林)是通过非竞争性的抑制HIV-l逆转录酶(RT)来抑制HIV-1复制。
Doravirine(多拉维林)不良反应:恶心,头晕,头痛,疲劳,腹泻,腹痛和做异常梦。
Doravirine(多拉维林)分子结构式如下:CAS:1338225-97-0英文名称:Doravirine中文名称:多拉维林二、Doravirine(多拉维林)合成路线三、Doravirine (多拉维林)合成检索总结报告(一)Doravirine (多拉维林)中间体3的合成合成方法实验步骤参考文献合成方法一To a round bottom flask charged with 3-bromo-5-chloro-phenol 2(5.20g,25.1mmol)and potassium carbonate (3.46g,25.1mmol)was added N-methylpyrrolidinone (25mL).To this suspension under N 2was added 2-chloro-3-fluoro-4-(trifluoromethyl)pyridine 1(5.00g,25.1mmol)and the reaction mixture was placed in an oil bath at 120o C.After 60minutes,the reaction mixture was allowed to cool to room temperature at which point,water (100mL)was added.The mixture was extracted with ethyl acetate (2x 100mL)and the combined organic fractions were washed with brine (3x 100mL),dried (MgSO 4),filtered and the solvent was evaporated under reduced pressure.The resulting residue was adsorbed onto silica gel and purified by column chromatography on a pre-packed silica gel Redi Sep 330gram column,eluting with 0-75%CH 2Cl 2in hexanes to yield the title compound 3.WO2009/67166;(2009);(A2)English A mixture of the 3-bromo-5-chlorophenol 2(3.74g;18.0合成方法二mmol),2-chloro-3-fluoro-4-(trifluoromethyl)pyridine1(3.00g;15.0mmol)and K2CO3(2.49g;18.0mmol)in NMP(15mL)was heated to120°C.for one hour,then cooled to room temperature.The mixture was then diluted with250mLEtOAc and washed with3×250mL1:1H2O:brine.Theorganic extracts were dried(Na2SO4)and concentrated invacuo.Purification by ISCO CombiFlash(120g column;load with toluene;100:0to0:100hexanes:CH2Cl2over40minutes)provided title compound3as a white solid.Repurification of the mixed fractions provided additionaltitle compound.US2011/245296;(2011);(A1)English(二)Doravirine(多拉维林)中间体4的合成合成方法实验步骤参考文献合成方法一To a suspension of3-(3-bromo-5-chlorophenoxy)-2-chloro-4-(trifluoromethyl)pyridine3(3.48g;8.99mmol)int-BuOH(36mL)was added KOH(1.51g;27.0mmol)andthe mixture was heated to75°C.overnight,at which point ayellow oily solid had precipitated from solution,and LCMSanalysis indicated complete conversion.The mixture wascooled to room temperature,and neutralized by the additionof50mL saturated aqueous NH4Cl.The mixture was dilutedwith50mL H2O,then extracted with2×100mL EtOAc.Thecombined organic extracts were dried(Na2SO4)andconcentrated in vacuo.Purification by ISCO CombiFlash(120g column;dry load;100:0to90:10CH2Cl2:MeOH over40minutes)provided the title com‐pound4as a fluffy white solid.US2011/245296;(2011);(A1)English合成方法二To a round bottom flask charged with3-(3-bromo-5-chlorophenoxy)-2-chloro-4-(trifluoromethyl)pyridine3(9.1g,25.3mmol)and potassium hydroxide(3.96g,70.5mmol)was added tert-butanol(100mL).This suspension wasplaced in an oil bath at75o C.After48hours,the reactionmixture was allowed to cool to room temperature and wasquenched with saturated aqueous ammonium chloride(50mL)and diluted with water(50mL).The mixture wasextracted with ethyl acetate(2x100mL)and the combinedWO2009/67166;(2009);(A2)English。
《美国抗癌药物化学合成速查》出版
佚名
【期刊名称】《中国医药工业杂志》
【年(卷),期】2009(40)3
【摘要】陈清奇教授主编的《美国抗癌药物化学合成速查》已由科学出版社于2009年2月出版,全书110万字,658页。
全书共介绍128个抗肿瘤药物。
内容包括:①美国所有已批准的抗肿瘤药物,其化学结构、用途和作用机理。
②每种药物有多少种制剂在美国上市和详细的产品信息,如商品名、剂型、剂量、处方药或非处方药、市场状态(上市或停止上市)、FDA产品申请号、批准时问和申报厂商等。
③化学药物的合成路线和相应的参考文献。
【总页数】1页(P212-212)
【关键词】科学出版社;抗癌药物;化学合成;美国;抗肿瘤药物;产品信息;非处方药;化学结构
【正文语种】中文
【中图分类】TQ464.7;G236
【相关文献】
1.药物合成反应在药物化学中的应用——以苯妥英的合成为例 [J], 刘冬梅;盛继文;张维芬;綦慧敏
2.药物化学合成实验中依达拉奉的合成方法改进 [J], 任江萌;谢贺新;刘慧
3.抗癌新药依维莫司的合成工艺优化和抗癌活性研究 [J], 王新利;兰燕芹;王尧
4.抗癌新药依维莫司的合成工艺优化和抗癌活性研究 [J], 王新利;兰燕芹;王尧
5.美国科学家合成抗癌分子 [J],
因版权原因,仅展示原文概要,查看原文内容请购买。
新药Vesatolimod(维沙莫德)合成检索总结报告
一、Vesatolimod(维沙莫德)简介
2020年3月10日公布了HIV治愈研究项目中Toll样受体7(TLR7)激动剂V esatolimod一项Ib期试验的结果。
数据显示:与安慰剂相比,vesatolimod的TLR7刺激作用与延长的病毒反弹时间、增强的免疫功能、完整HIV DNA水平的降低相关。
吉利德还公布了额外的临床前研究,评估在停止抗逆转录病毒治疗的情况下,Vesatolimod联合治疗方案实现病毒缓解的潜力。
Vesatolimod(维沙莫德)分子结构式如下:
英文名称:Vesatolimod
中文名称:维沙莫德
本文主要对Vesatolimod(维沙莫德)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Vesatolimod(维沙莫德)合成路线
三、Vesatolimod(维沙莫德)合成检索总结报告(一) Vesatolimod(维沙莫德)中间体3的合成方法一
(二) Vesatolimod(维沙莫德)中间体3的合成方法二
(三) Vesatolimod(维沙莫德)中间体5的合成。
药物Elbasvir(艾尔巴韦)合成检索总结报告一、Elbasvir(艾尔巴韦)简介Elbasvir(艾尔巴韦)于2016年1月在美国上市,主要用于治疗基因型1或4慢性丙肝病毒感染的成人患者。
Elbasvir(艾尔巴韦)常见的不良反应有疲劳、头痛和恶心等等。
Elbasvir(艾尔巴韦)分子结构式如下:CAS:1370468-36-2英文名称:Elbasvir中文名称:艾尔巴韦本文主要对Elbasvir(艾尔巴韦)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Elbasvir(艾尔巴韦)合成路线三、Elbasvir (艾尔巴韦)合成检索总结报告(一)Elbasvir (艾尔巴韦)中间体3的合成合成方法实验步骤参考文献操作方法一Into a 1000mL flask were charged 2-(2-bromo-5-chlorophenyl)acetic acid 1(60g,242mmo1)and trifluoromethanesulfonic acid (1.1kg).The mixture was allowed to stir for 10minutes,then 3-chlorophenol 2(27g,211mmol)was added.The reaction was heated to 55o C and allowed to stir at this temperature for about 15hours.The reaction mixture was then cooled to room temperature and poured into 3kg of ice-water.The suspension formed was allowed to stir for 30minutes and then filtered.The solid was collected and washed with water (300mL×3).The solid was dissolved in ethyl acetate (1000mL was discard)and the collected organic layer was dried with Na 2SO 4,filtered and concentrated in vacuo to provide compound 3(82g,crude)as a solid.It wasused directly to the next step.WO2016/4899;(2016);(A1)English;Organic Process Research and Development ;vol.21;nb.10;(2017);p.1547–1556.(二)Elbasvir (艾尔巴韦)中间体4的合成合成方法实验步骤参考文献操作方法一A solution of ammonia (7M in methanol)was added to compound 3(10g)and the resulting reaction was allowed to stir at room temperature for 20hours.The slurry formed was filtered and the collected solid was washed with methanol then dried in vacuo to provide compound 4as a solid 8.6g,86%yield.WO2016/4899;(2016);(A1)English 操作方法二Into a 1000ml round bottom flask was placed 2-(2-bromo-5-chlorophenyl)-1-(4-chloro-2-hydroxyphenyl)et hanone 3(82.0g,crude),ammonia in MeOH (400ml,ca.9N).The resulting mixture was stirred at room temperature overnight.The solid was filtered and washed with MeOH (60ml ×2),and then dried under reduced pressure to give 2-(2-(2-bromo-5-chlorophenyl)-1-iminoethyl)-5-Organic Process Research and Development ;vol.21;nb.10;(2017);p.1547–1556.。
药物Letermovir(莱特莫韦)合成检索总结报告
一、Letermovir(莱特莫韦)简介
Letermovir(莱特莫韦)于2017年11月在美国上市,主要用于治疗接受异基因造血干细胞移植后巨细胞病毒血清呈阳性的成人患者,可预防CMV感染。
Letermovir(莱特莫韦)是非核苷CMV抑制剂,常见的不良反应有恶心、腹泻、呕吐、外周水肿、咳嗽、头痛、疲劳和腹痛。
Letermovir(莱特莫韦)分子结构式如下:
CAS:917389-32-3
英文名称:Letermovir
中文名称:莱特莫韦
本文主要对Letermovir(莱特莫韦)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Letermovir(莱特莫韦)合成路线
三、Letermovir (莱特莫韦)合成检索总结报告
(一)Letermovir (莱特莫韦)中间体3
的合成合成方法
实验步骤参考文献操作方法
一To a
degassed solution of 2-bromo-6-fluoroaniline 1(1,99.5g,0.524mol),methyl acrylate 2(95.0mL,1.05mol),Chloro[(tri-tert-butylphosphine)-2-(2-aminobiphenyl)]palladium(II)(0.537g,1.05mmol)in isopropyl acetate (796mL),was added degassed N,N-dicyclohexylmethylamine (135mL,0.628mol).The resulting reaction was heated to 80°C and allowed to stir at this temperature for 5hours.The resulting slurry was cooled to 20°C and filtered.The filtrate was washed with 1M citric acid to provide a solution that contained compound 3(99.3g,97%assay yield)in isopropyl acrylate,which was used without further purification.WO2015/88931;(2015);(A1)English (二)Letermovir (莱特莫韦)中间体5的合成合成方法
实验步骤参考文献操作方法
一To a solution of compound 3(48.8g,0.250mol)in 683mL of isopropyl acetate was added 244mL of water,followed by di-sodium hydrogen phosphate (53.2g,0.375mol).To the resulting solution was added phenyl chloroformate 4(39.2mL,0.313mol)dropwise over 30minutes.The resulting reaction was heated to 30°C and allowed to stir at this temperature for 5hours for 4hours and then was heated to 60°C and allowed to stir at this temperature for 5hours for an additional 2hours to remove excess phenyl chloroformate.An additional 293mL of isopropyl acetate was then added and the reaction mixture was allowed to stir
WO2015/88931;(2015);(A1)English
at room temperature until the solids completely dissolved
into solution.The resulting reaction mixture was transferred to a separatory funnel and the organic phase was washed with 98mL of water and collected to provide a solution of compound 5in isopropyl acetate,which was used without further purification.操作方法
二To a stirred crude i -PrOAc solution of acrylate 3(15.9kg,8.2wt %15,6.66mol)was added H 2O (6.50L)and Na 2HPO 4(1.42kg,10.0mol)followed by phenyl chloroformate 4(1.04L,8.33mol)dropwise over 30min.The reaction mixture was stirred at RT for 12h before being heated to 60°C and stirred for a further 2h.The mixture was then diluted with i -PrOAc (7.00L)and held at 60°C until all solids were dissolved.The aqueous phase was then separated and the organics washed with H 2O (2.60L)before being cooled to RT to afford a crude i-PrOAc slurry of carbamate 5(ca.95%assay yield)which was used directly without further purifification.For the purposes of characterization,a sample of pure carbamate 5was isolated by crystallization from a sample of the crude solution diluted with heptane.Mp 154−158°anic Process Research and Development ;vol.20;nb.6;(2016);p.1097–1103.
(三)Letermovir (莱特莫韦)中间体7的合成方法一合成方法
实验步骤参考文献操作方法
一
A solution of compound 5(79.0g,0.250mol),2-methoxy-5-(trifluoromethyl)aniline 6(52.7g,0.276mol),and 4-dimethylaminopyridine (0.92g,0.0075mol)in isopropyl acetate (780mL)was heated to reflux and allowed to stir at this temperature for 5hours.The resulting slurry was cooled to 20°C,then allowed to stir at this temperature for for two hours at this temperature,then filtered.The collected filter cake was dried in vacuo to provide compound 7(95.0g,0.230mol)as a white solid,which was used without further purification.WO2015/88931;(2015);(A1)English 操作方法To a stirred crude i-PrOAc solution of carbamate 5(<6.66mol)was added 2-methoxy-5-(triflfluoromethyl)aniline 6(1.40kg,7.33mol).The slurry was diluted with i -PrOAc,heated to 40−45°C,and a constant volume distillation carried out to azeotropically dry the mixture (KF <400ppm).
Organic Process Research and Development ;。