第三章势能面、构型优化 与构象搜索
- 格式:pdf
- 大小:2.34 MB
- 文档页数:28

计算机辅助药物设计知到章节测试答案智慧树2023年最新暨南大学第一章测试1.电影《我不是药神》中的神药“格列宁”是用于治疗参考答案:白血病2.计算机辅助药物设计的应用可以加快靶点发现的速度,参考答案:提高靶点发现的准确度3.目前计算机辅助药物设计存在的问题参考答案:药物体内转运、代谢和体内毒副作用问题;受体-配体相互作用的方式问题;设计的分子能否进行化学合成;蛋白质结构三维结构的真实性和可用性问题4.计算机辅助药物设计是应用量子力学、分子动力学、构效关系等基础理论数据研究药物对酶、受体等作用的药效模型, 从而达到药物设计之目的参考答案:对5.目前已经可以纯粹依靠计算机设计出一个分子即成为新药。
参考答案:错6.先导化合物是通过各种途径、方法、手段获得的具有某种药理学或生物学活性的化合物。
其不一定为实用的优良药物,可能因其药效不强、特异性不高、毒性较大等而不能直接药用,需要进行结构修饰和改造,使之成为实用的高效、低毒、可控的优良药物,这一过程称为先导化合物的优化.参考答案:对第二章测试1.地西泮具有以下哪类结构母核参考答案:苯并二氮卓类2.阿司匹林解热作用机制是参考答案:抑制环氧酶,减少PG合成3.地西泮何处的水解开环是可逆的参考答案:4、5位4.药物作用的靶点包括参考答案:离子通道;受体;酶;内源性小分子物质5.受体都有其内源性配体,如参考答案:激素;自体活性物质;神经递质6.受体具有哪些特征参考答案:特异性;可逆性;高亲和力;饱和性7.药物作用的特异性,取决于药物分子也就是配体, 与其受体结合的专一性参考答案:对8.现代药物发现策略也面临诸多挑战,如所选“靶点”的成药性问题及可能发生的“脱靶效应”。
参考答案:对9.体内能与受体特异性结合的物质,称为配体或配基参考答案:对10.能够与药物结合产生相互作用,发动细胞反应的大分子或大分子复合物的物质称为受体参考答案:对第三章测试1.建立QSAR模型,最常用的就是参考答案:偏最小二乘法2.下列哪些是定量构效关系CoMFA方法用到的参数参考答案:指示变量;结构参数;立体参数;电性参数3.分子场分析法假定配基与受体的结合是共价的,两者的亲和力或配基的生物活性与配基分子周围的空间立体场或静电场有关。



构型构象分析范文构型构象分析是一种解析和理解分子结构的方法,它通过分析分子内部和外部的几何排列,来推断分子的物理性质和化学行为。
该方法可应用于有机分子、配位化合物、金属有机化合物、配位聚合物、化学反应的过渡态等多种体系。
构型构象分析通常包括构型优化、构型和构象分析三个步骤。
首先是构型优化。
构型优化旨在找到分子能量最低的构象。
该步骤通常采用分子力场方法,以计算化学和物理量,如键长、键角、扭转角、电子结构等,优化分子构型。
分子力场方法基于近似势能面,在计算中使用原子间作用力场参数来模拟分子相互作用。
此外,还可以使用量子力学计算方法,如密度泛函理论,通过解耦化学键的电子波函数,计算分子结构。
其次是构型。
构型用于寻找构象空间中的所有可能构象。
构象空间是指分子在给定的化学方式下能够实现的所有三维排列方式。
构象可以通过多种方法实现,如蒙特卡洛方法、分子动力学模拟、以及遗传算法等。
这些方法可以在不同的温度下模拟分子的运动,并在中使用能量、几何约束等条件来探索构象空间。
通过构象,可以获得一系列可能的构象,并将它们用于后续的构象分析。
最后是构象分析。
构象分析是对构象进行定量和定性的分析,并与实验数据进行比较和验证。
构象分析通常涉及几何参数、能量特征、键键相互作用等方面的分析。
几何参数分析可以计算化学键的长度、角度和扭转角等,并与实验数据进行比较来验证构象的准确性。
能量特征分析可以研究构象的相对稳定性,通过比较构象的能量差异来得出稳定构象。
键键相互作用的分析可以研究构象对分子性质和化学行为的影响,进一步理解构象对于分子的作用。
在实际应用中,构型构象分析在药物设计、催化反应、材料科学等领域具有重要的应用价值。
例如,在药物设计中,通过对药物分子的构型构象进行分析,可以预测分子的活性位点、药效和毒性,为药物研发提供有价值的信息。
在催化反应中,构型构象分析可以研究催化剂的几何结构对反应速率和选择性的影响,指导催化剂的设计和合成。
![[高一理化生]第3章本章优化总结PPT课件](https://img.taocdn.com/s1/m/ac9c175ebceb19e8b9f6ba27.png)
实验分⼦构型优化实验⼀分⼦构型优化⼀、⽬的要求1. 了解Gaussian程序中优化分⼦结构的基本原理和流程。
2. 掌握优化分⼦结构的计算技术及判断优化是否正常完成的标准。
3. 学会查看结果⽂件并能对简单出错信息进⾏处理。
⼆、基本原理寻找分⼦的平衡⼏何构型是计算化学中最常见最普遍的应⽤。
学过结构化学,我们知道,分⼦的结构决定其性质。
在实际的实验过程中,可能有很多原因,使我们很难观测到分⼦的稳定结构,如存在寿命很短的中间体、过渡态、或者是混合物难以分离、或者因为不稳定容易分解等很多因素,使得实验上测到分⼦稳定构型的可能性很⼩,这时我们就可以借助计算化学来帮忙预测。
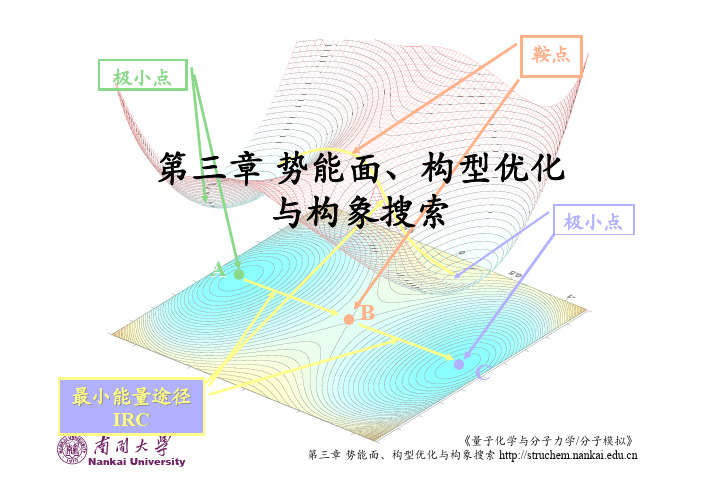
2.1 势能⾯在进⾏分⼦结构优化前,⾸先我们需要了解势能⾯(Potential Energy Surface,PES)的概念。
根据Born-Oppenheimer近似,分⼦基态的能量可以看作只是核坐标的函数,体系能量的变化可以看成是在⼀个多维⾯上的运动,这个多维⾯就是势能⾯。
势能⾯是⼀个超平⾯,由势能对全部原⼦的可能位置构成,全部原⼦的位置可⽤3N-6个坐标来表⽰(双原⼦分⼦,独⽴坐标数为1)。
其中,在直⾓坐标系中,N指的是原⼦数,3N是指直⾓坐标数,描述平动坐标的数为3,描述转动坐标的数为3,独⽴的坐标数为3N-6;在内坐标系中,内坐标⾃由度的数⽬为3N-6。
具体来讲,在不分解的前提下,分⼦可以有很多个可能的构型,每个构型都有⼀个能量值,所有这些可能的结构所对应的能量值的图形表⽰就是⼀个势能⾯。
势能⾯描述的是分⼦结构和其能量之间的关系,以能量和坐标作图。
势能⾯上的每⼀个点对应⼀个结构。
势能⾯上的点最令我们感兴趣的是势能对坐标⼀阶偏导数为零的点,即梯度(gradient)都为零。
势能对坐标⼀阶偏导数对应着⼒,因此处于势能⾯上这样的点所受到的⼒为零,这样的点称为驻点(stationary point)。
驻点分为三种,极⼩点(Minimum)、极⼤点(Maximum)和鞍点(saddle point),如图2-1所⽰。