YYT0287-2017标准与医疗器械法规对照表
- 格式:xls
- 大小:56.00 KB
- 文档页数:24


YY/T0287-2017 idt ISO13485:2016《医疗器械质量管理体系用于法规的要求》标准解读(一)2017年02月04日发布一、概述1.ISO13485标准的简要回顾ISO13485标准已经经历了两个版本,1996年ISO发布了ISO13485:1996《质量体系—医疗器械—ISO9001应用的专用要求》标准,该标准不是独立标准,而是要和ISO9001:1994标准联合使用的标准。
2003年ISO/TC210修订1996版ISO13485标准后,发布了ISO13485:2003《医疗器械质量管理体系用于法规的要求》标准,该标准是专用于医疗器械领域的独立标准。
目前ISO/TC210已正式于2016年3月1日发布实施第三版的ISO13485标准。
国家食品药品监督管理总局及时将该标准转化为YY/T0287-2017 idt ISO13485:2016《医疗器械质量管理体系用于法规的要求》标准发布实施。
ISO13485标准是应用于医疗器械领域的质量管理体系标准,该标准突出关注医疗器械的安全有效,强调组织提供的医疗器械要满足顾客要求和法规要求。
由于ISO13485标准的宗旨和医疗器械法规的目标高度契合,与医疗器械产业界及社会公众的期望完全一致,因此ISO13485标准一经发布,就得到全球医疗器械产业界、监管部门及社会的高度重视及广泛认可。
很多国家将ISO13485标准转化为本国标准,在医疗器械领域贯彻实施。
我国政府高度重视ISO13485标准。
医疗器械监管部门积极跟踪ISO13485标准的制修订过程,分别在1996年和2003年ISO13485标准发布后即等同采用转化为行业标准YY/T 0287-1996和YY/T 0287-2003标准,确保我国行业标准发布和国际标准保持同步。
医疗器械监管部门在制定相关医疗器械法规时也引用和借鉴了ISO13485标准的要求。
在政府和市场推动下,ISO13485标准的理念、原则和方法在我国医疗器械产业界得到迅速传播和广泛应用,并取得巨大成功。

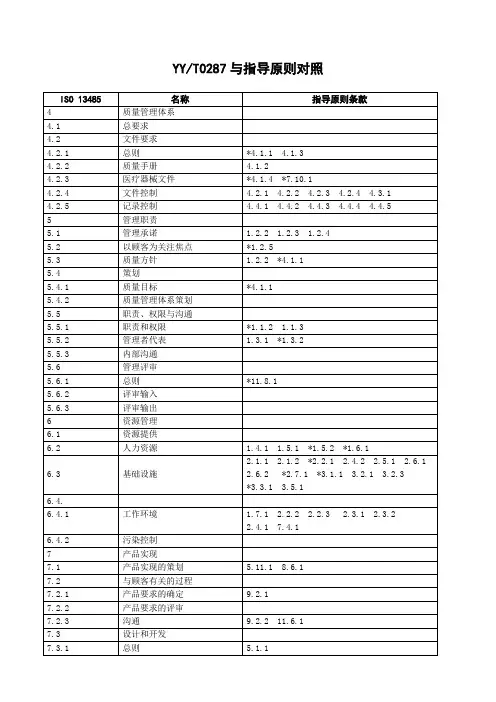

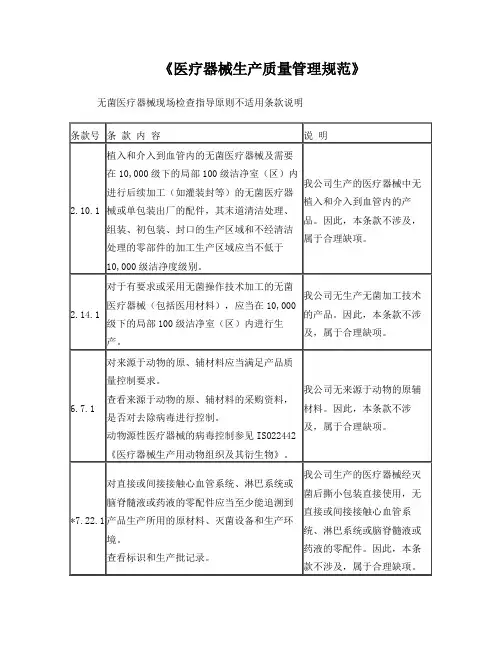
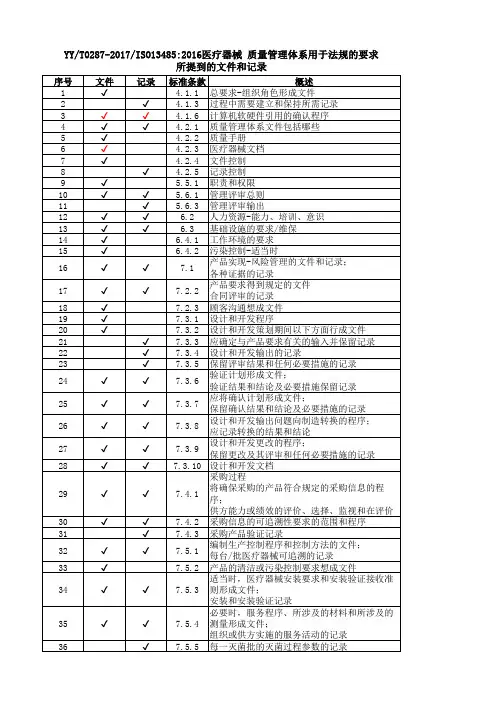

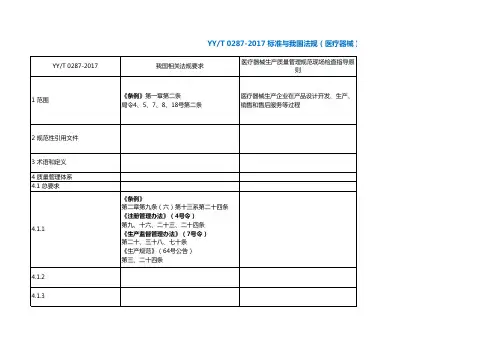
YY/T 0287-2017 标准与我国法规(医疗器械)对
8.3.2 交付前发现不合格品的响应措施《生产规范》64号公告第六十八条
10.1.1
*10.2.1
8.3.3 交付后发现不合格品的响应措施《生产规范》64号公告第六十九条
10.3.1
*11.5.1 对已交付的不合格品的响应措施及
向监管机构报告的具体要求
8.3.4 返工《生产规范》64号公告第七十条10.4.1 10.4.2
8.4 数据分析《生产规范》64号公告第七十三条11.3.1 明确收集的质量数据还包括不良事件信息
8.5 改进
8.5.1 总则《生产规范》第七十四条
8.5.2 纠正措施《生产规范》第七十四条11.4.1 8.5.3 预防措施《生产规范》第七十四条11.4.2
械)对应关系。
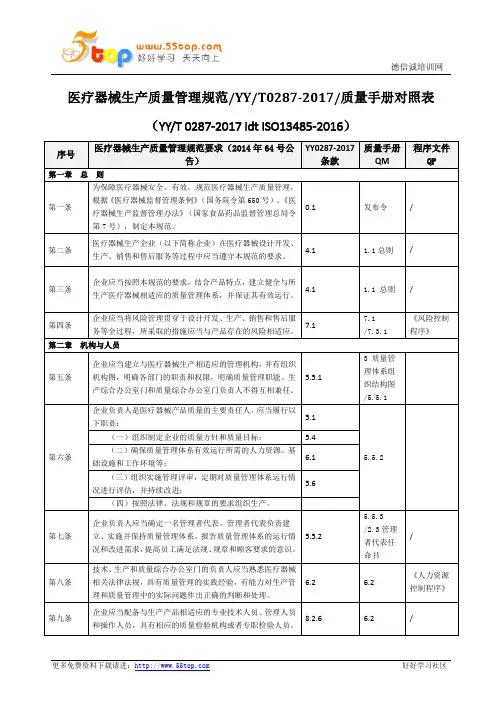
医疗器械生产质量管理规范/YY/T0287-2017/质量手册对照表(YY/T 0287-2017 idt ISO13485-2016)序号医疗器械生产质量管理规范要求(2014年64号公告)YY0287-2017条款质量手册QM程序文件QP第一章总则第一条为保障医疗器械安全、有效,规范医疗器械生产质量管理,根据《医疗器械监督管理条例》(国务院令第650号)、《医疗器械生产监督管理办法》(国家食品药品监督管理总局令第7号),制定本规范。
0.1 发布令/第二条医疗器械生产企业(以下简称企业)在医疗器械设计开发、生产、销售和售后服务等过程中应当遵守本规范的要求。
4.1 1.1总则/第三条企业应当按照本规范的要求,结合产品特点,建立健全与所生产医疗器械相适应的质量管理体系,并保证其有效运行。
4.1 1.1 总则/第四条企业应当将风险管理贯穿于设计开发、生产、销售和售后服务等全过程,所采取的措施应当与产品存在的风险相适应。
7.17.1/7.3.1《风险控制程序》第二章机构与人员第五条企业应当建立与医疗器械生产相适应的管理机构,并有组织机构图,明确各部门的职责和权限,明确质量管理职能。
生产综合办公室门和质量综合办公室门负责人不得互相兼任。
5.5.13 质量管理体系组织结构图/5.5.1第六条企业负责人是医疗器械产品质量的主要责任人,应当履行以下职责:5.15.5.2(一)组织制定企业的质量方针和质量目标; 5.4(二)确保质量管理体系有效运行所需的人力资源、基础设施和工作环境等;6.1(三)组织实施管理评审,定期对质量管理体系运行情况进行评估,并持续改进;5.6(四)按照法律、法规和规章的要求组织生产。
第七条企业负责人应当确定一名管理者代表。
管理者代表负责建立、实施并保持质量管理体系,报告质量管理体系的运行情况和改进需求,提高员工满足法规、规章和顾客要求的意识。
5.5.25.5.3/2.3管理者代表任命书/第八条技术、生产和质量综合办公室门的负责人应当熟悉医疗器械相关法律法规,具有质量管理的实践经验,有能力对生产管理和质量管理中的实际问题作出正确的判断和处理。
文件编号:CMHZ-SC-01控制状态:受控 非受控□版本号:000发放编号:CMHZ-2017-001质量手册编制:审核:批准:2017年9月29日发布 2017年9月29日实施XXX公司目录1.范围 (6)1.1总则 (6)1.2删减和不适用说明 (6)1.2.1 YY/T 0287-2017 idt ISO13485-2016不适用条款说明: (6)1.2.2医疗器械生产质量管理规范不适用条款说明: (6)1.3引用的法规和标准 (7)1.4质量手册的管理 (8)1.5质量方针与质量目标 (9)质量方针: (9)质量目标: (9)2 企业概况 (10)2.1修改页 (11)2.2颁布令 (12)2.3管理者代表任命书 (13)2.4公司管理架构图 (13)3 质量管理体系组织结构图 (14)4.质量管理体系 (15)4.1总要求 (15)4.2文件要求 (16)4.2.1总则 (16)4.2.2质量手册 (18)4.2.3医疗器械文档 (18)4.2.4文件控制 (18)4.2.5记录控制 (19)4.3支持性文件 (20)5.管理职责 (20)5.1管理承诺 (20)5.2以客户为关注焦点 (20)5.3质量方针 (20)5.4策划 (21)5.4.1质量目标 (21)5.4.2质量管理体系策划 (21)5.5职责、职权与沟通 (22)5.5.1职责与权限 (22)5.5.2总经理 (23)5.5.3管理者代表 (23)5.5.4综合办公室 (24)5.5.5品保部: (25)5.5.6技术部 (26)5.5.7运营部 (26)5.5.8市场部 (27)5.5.9销售部 (28)5.5.10内部沟通 (28)5.6管理评审 (29)5.6.1总则 (29)5.6.2评审输入 (29)5.6.3评审输出 (29)5.7支持性文件 (30)6.资源管理 (31)6.1提供资源 (31)6.2人力资源 (31)6.3基础设施 (31)6.4工作环境和污染的控制 (32)6.4.1工作环境 (32)6.5支持文件 (32)7.产品实现 (33)7.1产品实现的策划 (33)7.1.1 适用范围 (33)7.1.2 策划内容 (33)7.1.3 质量计划 (33)7.2与顾客有关的过程 (34)7.2.1产品要求的确定 (34)7.2.2产品要求的评审 (34)7.2.3沟通 (35)7.3设计和开发 (35)7.3.1总则 (35)7.3.2设计和开发策划 (35)7.3.3设计和开发输入 (35)7.3.4设计和开发输出 (36)7.3.5设计和开发评审 (36)7.3.6设计和开发验证 (36)7.3.7设计和开发确认 (37)7.3.8设计和开发的转换 (37)7.3.9设计和开发更改的控制 (37)7.3.10设计和开发文档 (37)7.3.11风险管理 (37)7.4采购 (38)7.4.1采购过程 (38)7.4.2采购信息 (38)7.4.3采购产品的验证 (39)7.5生产和服务提供 (39)7.5.1生产和服务提供控制 (39)7.5.2产品的清洁 (39)7.5.4服务活动 (40)7.5.5无菌医疗器械的专用要求 (40)7.5.6生产和服务提供过程的确认 (40)7.5.7灭菌过程和无菌屏障系统确认的专用要求 (40)7.5.8标识 (41)7.5.9可追溯性 (41)7.5.10顾客财产 (42)7.5.11产品防护 (42)7.6监视和测量设备的控制 (42)7.7相关文件 (43)8测量、分析和改进 (43)8.1总则 (43)8.2监视和测量 (43)8.2.1反馈 (43)8.2.2投诉处置 (44)8.2.3向监管机构报告 (44)8.2.4内部审核 (44)8.2.5过程监视和测量 (45)8.2.6产品监视和测量 (45)8.3不合格产品控制 (46)8.3.1总则 (46)8.3.2交付前发现不合格品的相应措施 (46)8.3.3交付后发现的不合格品的响应措施 (46)8.3.4返工 (46)8.4数据分析 (46)8.5改进 (47)8.5.1总则 (47)8.5.2纠正措施 (47)8.5.3预防措施 (48)附件一程序文件清单 (49)附件二职能分配表 (50)附件三医疗器械生产质量管理规范/YY/T0287-2017/质量手册对照表 (51)1.范围1.1总则本手册是依据YY/T0287-2017/ISO13485:2016《医疗器械质量管理体系用于法规的要求》和GB/T 19001-2016/ISO9001:2015《质量管理体系要求》标准制定。
总局关于批准发布YY/T 0287—2017《医疗器械质量管理体系用于法规的要求》医疗器械行业标准的公告(2017年第11
号)
YY/T 0287—2017《医疗器械质量管理体系用于法规的要求》医疗器械行业标准已经审定通过,现予以公布。
该标准自2017年5月1日起实施。
标准编号、名称及适用范围见附件。
特此公告。
附件:YY/T 0287—2017《医疗器械质量管理体系用于法规的要求》医疗器械行业标准编号、名称及适用范围
食品药品监管总局
2017年1月19日
附件
YY/T0287—2017《医疗器械质量管理体系用于法规的要求》医疗器械行业标准
编号、名称及适用范围
YY/T 0287—2017《医疗器械质量管理体系用于法规的要求》
本标准规定了需要证实其有能力提供持续满足顾客要求和适用的法规要求的医疗器械和相关服务的组织的质量管理体系要求。
本标准适用于涉及医疗器械生命周期的产业链的各类组织,即医疗器械的设计开发和生产企业、经营企业、物流企业、科研机构、维修服务公司、安装公司,以及向医疗器械组织提供产品的供方或其他外部方(如提供原材料、组件、部件、医疗器械、灭菌服务、校准服务、流通服务、维护服务等组织)。
本标准代替YY/T 0287—2003《医疗器械质量管理体系用于法规的要求》。
RR/T0287:2017医疗器械质量管理体系质量手册2017年11月21日发布2017年11月21日实施目录0.1、前言及引言0.2、管理者代表任命书0.3、质量手册发布令20.4、质量手册发布令1.范围1.1总则1.2应用2.规范性引用文件3.术语和定义4.质量管理体系4.1总要求4.2文件要求4.2.1总则4.2.2质量手册4.2.3医疗器械文件4.2.3文件控制4.2.4记录控制5.管理职责5.1管理承诺5.2以顾客为关注焦点5.3质量方针5.4策划5.4.1质量目标5.4.2质量管理体系的策划5.5职责、权限和沟通5.5.1职责和权限5.5.2管理者代表5.6管理评审5.6.1总则5.6.2评审输入5.6.3评审输出6.资源管理6.1资源提供6.2人力资源6.3基础设施6.4工作环境和污染控制7.产品实现7.1产品实现的策划7.2与顾客有关的过程7.2.1与产品有关的要求的确定7.2.2与产品有关要求的评审7.2.3沟通7.3设计和开发7.3.1总则7.3.2设计和开发策划7.3.3设计和开发输入7.3.4设计和开发输出7.3.5设计和开发评审7.3.6设计和开发验证7.3.7设计和开发确认7.3.8设计和开发转换7.3.9设计和开发更改的控制7.3.10设计和开发文档7.4采购7.4.1采购过程7.4.2采购信息7.4.3采购产品的验证7.5生产和服务提供7.5.1生产和服务提供的控制7.5.2产品的清洁7.5.3安装活动7.5.4服务活动7.5.5无菌医疗器械的专用要求7.5.6生产和服务提供过程的确认7.5.7灭菌过程和无菌屏障系统确认的专用要求7.5.8标识7.5.9可追溯性7.5.10顾客财产7.5.11产品防护7.6监视和测量设备的控制8.测量、分析和改进8.1总则8.2监视和测量8.2.1反馈8.2.2抱怨处理8.2.3向监管机构报告8.2.4内部审核8.2.5过程的监视和测量8.2.6产品的监视和测量8.3不合格品控制8.3.1总则8.3.2交付前发现不合格品的响应措施8.3.3交付后发现的不合格品的响应措施8.4数据分析8.5改进8.5.1总则8.5.2纠正措施8.5.3预防措施9.MDD要求9.1总则9.2适用范围9.3职责9.4程序要求附录A公司质量目标附录B各部门目标分解附录C职责分配表附录D程序文件清单附录E组织架构图附录FRR/T0287-2017和GB/T19001-2016对应关系表0.2、前言及简述本文件为RR市RRRR有限公司之质量手册,详述本公司为符合顾客之要求所提供之质量、保证。
质量手册(ISO13485-2016/ YYT-0287-2017)1、目的范围1.1 目的公司依据《医疗器械生产质量管理规范》、《规范附录无菌医疗器械》、YY/T 0287-2017 idt ISO 13485:2016《医疗器械质量管理体系用于法规要求》、GB/T19001-2016 idt ISO9001:2015是指《质量管理体系要求》及相关法律、法规、规章和标准,结合公司的实际,建立并实施质量管理体系,以达到以下目的:A)证实具有能稳定地提供满足顾客要求和适用于本公司产品的医疗器械和相关服务的法规要求的能力;B)通过体系的有效应用,包括体系持续改进的过程的有效应用以及保证符合顾客与适用的法律法规要求,旨在增强顾客满意;C)实施、保持并改进质量管理体系;D)使自己确信能符合所申明的质量方针,并向外界展示这种符合性;E)寻求外部组织对其质量体系的审核、认证、注册,进行自我鉴定和自我申明。
1.2范围1.2.1 本手册适用于本公司生产的***********、***********等产品的设计、开发、生产和服务过程。
1.2.2 本手册适用于领导层(总经理、副总经理)、销售部、研发部、质量部、生产部、采购部和售后部、行政部、人力资源部及与产品质量有关的过程、活动和人员;1.2.3本手册适用于内部质量管理及对外提供质量保证(第二方审核或第三方认证)。
1.2.4按法规要求对第七章的不适用部分作了删减:删减理由说明如下:本公司的灭菌过程完全交由第三方完成,因此,在本质量管理体系中将《YY/T 0287-2017医疗器械质量管理体系用于法规的要求》标准中的“7.5.5无菌医疗器械的专用要求”的章节予以删减。
基于同样的理由将《规范附录无菌医疗器械》要求中的“2.6.9”条款予以删减。
本公司无植入和介入到血管内的无菌医疗器械,因此,在本质量管理体系中将《YY/T 0287-2017医疗器械质量管理体系用于法规的要求》标准中的“7.5.9.2植入性医疗器械的专项要求”的章节予以删减。