保护性碱基集锦
- 格式:pdf
- 大小:53.35 KB
- 文档页数:4


保护碱基表全集文档(可以直接使用,可编辑实用优质文档,欢迎下载)
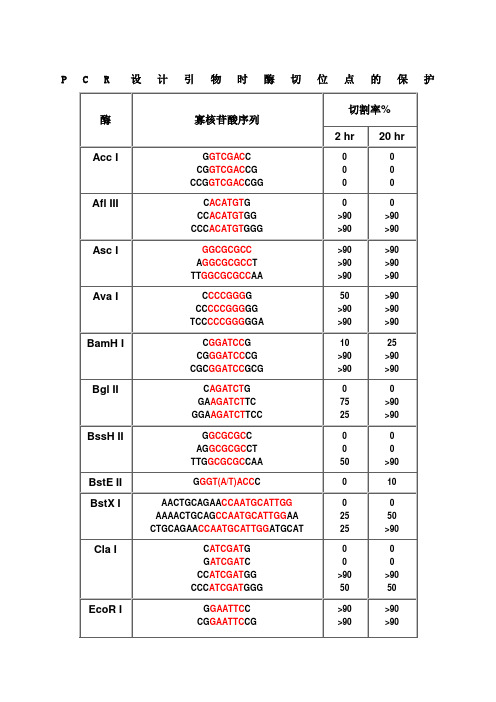
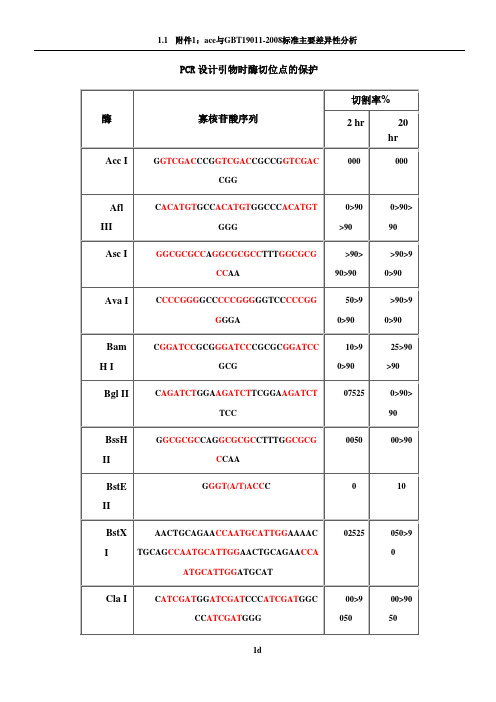
不同内切酶对识别位点以外最少保护碱基数目的要求PCR设计引物时酶切位点的保护碱基表1
PCR设计引物时酶切位点的保护碱基表2
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的,见附表。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
保护碱基列表
PCR设计引物时酶切位点的保护
注释:
1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
2.切割率:正确识别并酶切的效率
3。
加保护碱基时最好选用切割率高时加的相应碱基。


各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments (oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同 buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。
常用酶切位点的保护性碱基Enzyme OligoSequenceChain %Cleavage Length 2hr 20hrAccIG GTCGAC C 8 0 0 CG GTCGAC CG 10 0 0 CCG GTCGAC CGG 12 0 0AflIIIC ACATGT G 8 0 0 CC ACATGT GG 10 >90 >90 CCC ACATGT GGG 12 >90 >90AscIGGCGCGCC 8 >90 >90 A GGCGCGCC T 10 >90 >90 TT GGCGCGCC AA 12 >90 >90AvaIC CCCGGG G 8 50 >90 CC CCCGGG GG 10 >90 >90 TCC CCCGGG GGA 12 >90 >90BamHIC GGATCC G 8 10 25 CG GGATCC CG 10 >90 >90 CGC GGATCC GCG 12 >90 >90BglIIC AGATCT G 8 0 0 GA AGATCT TC 10 75 >90 GGA AGATCT TCC 12 25 >90BssHIIG GCGCGC C 8 0 0 AG GCGCGC CT 10 0 0 TTG GCGCGC CAA 12 50 >90BstEII G GGT(A/T)ACC C 9 0 10BstXIAACTGCAGAA CCAATGCATTGG22 0 0 AAAACTGCAG CCAATGCATTGG AA 24 25 50 CTGCAGAA CCAATGCATTGG ATGCAT 27 25 >90ClaIC ATCGAT G 8 0 0G ATCGAT C 8 0 0 CC ATCGAT GG 10 >90 >90 CCC ATCGAT GGG 12 50 50EcoRIG GAATTC C 8 >90 >90 CG GAATTC CG 10 >90 >90 CCG GAATTC CGG 12 >90 >90HaeIIIGG GGCC CC 8 >90 >90 AGC GGCC GCT 10 >90 >90TTGC GGCC GCAA 12 >90 >90HindIIIC AAGCTT G 8 0 0 CC AAGCTT GG 10 0 0 CCC AAGCTT GGG 12 10 75KpnIG GGTACC C 8 0 0 GG GGTACC CC 10 >90 >90 CGG GGTACC CCG 12 >90 >90MluIG ACGCGT C 8 0 0 CG ACGCGT CG 10 25 50NcoIC CCATGG G 8 0 0 CATG CCATGG CATG 14 50 75NdeIC CATATG G 8 0 0CC CATATG GG 10 0 0CGC CATATG GCG 12 0 0 GGGTTT CATATG AAACCC 18 0 0 GGAATTC CATATG GAATTCC 20 75 >90 GGGAATTC CATATG GAATTCCC 22 75 >90NheIG GCTAGC C 8 0 0 CG GCTAGC CG 10 10 25 CTA GCTAGC TAG 12 10 50NotITT GCGGCCGC AA 12 0 0 ATTT GCGGCCGC TTTA 16 10 10 AAATAT GCGGCCGC TATAAA 20 10 10 ATAAGAAT GCGGCCGC TAAACTAT 24 25 90 AAGGAAAAAA GCGGCCGC AAAAGGAAAA 28 25 >90NsiITGC ATGCAT GCA 12 10 >90 CCA ATGCAT TGGTTCTGCAGTT 22 >90 >90PacITTAATTAA 8 0 0 G TTAATTAA C 10 0 25 CC TTAATTAA GG 12 0 >90PmeIGTTTAAAC 8 0 0G GTTTAAAC C 10 0 25GG GTTTAAAC CC 12 0 50 AGCTTT GTTTAAAC GGCGCGCCGG 24 75 >90PstIG CTGCAG C 8 0 0TGCA CTGCAG TGCA 14 10 10 AA CTGCAG AACCAATGCATTGG 22 >90 >90 AAAA CTGCAG CCAATGCATTGGAA 24 >90 >90 CTGCAG AACCAATGCATTGGATGCAT26 0 0PvuIC CGATCG G 8 0 0 AT CGATCG AT 10 10 25 TCG CGATCG CGA 12 0 10SacI C GAGCTC G 8 10 10SacIIG CCGCGG C 8 0 0 TCC CCGCGG GGA 12 50 >90SalIGTCGAC GTCAAAAGGCCATAGCGGCCGC28 0 0 GC GTCGAC GTCTTGGCCATAGCGGCCGCGG 30 10 50 ACGC GTCGAC GTCGGCCATAGCGGCCGCGGAA 32 10 75ScaIG AGTACT C 8 10 25 AAA AGTACT TTT 12 75 75SmaICCCGGG 6 0 10C CCCGGG G 8 0 10 CC CCCGGG GG 10 10 50 TCC CCCGGG GGA 12 >90 >90SpeIG ACTAGT C 8 10 >90GG ACTAGT CC 10 10 >90 CGG ACTAGT CCG 12 0 50 CTAG ACTAGT CTAG 14 0 50SphIG GCATGC C 8 0 0 CAT GCATGC ATG 12 0 25 ACAT GCATGC ATGT 14 10 50StuIA AGGCCT T 8 >90 >90 GA AGGCCT TC 10 >90 >90 AAA AGGCCT TTT 12 >90 >90XbaIC TCTAGA G 8 0 0GC TCTAGA GC 10 >90 >90 TGC TCTAGA GCA 12 75 >90 CTAG TCTAGA CTAG 14 75 >90XhoIC CTCGAG G 8 0 0 CC CTCGAG GG 10 10 25 CCG CTCGAG CGG 12 10 75XmaIC CCCGGG G 8 0 0CC CCCGGG GG 10 25 75 CCC CCCGGG GGG 12 50 >90 TCCC CCCGGG GGGA 14 >90 >90。
克隆PCR产物的方法之一,是在PCR产物两端设计一定的限制酶切位点,经酶切后克隆至用相同酶切的载体中。
但实验证明,大多数限制酶对裸露的酶切位点不能切断。
必须在酶切位点旁边加上一个至几个保护碱基,才能使所定的限制酶对其识别位点进行有效切断。
有研究者使用了15种限制酶,分别比较了各种限制酶在其酶切位点旁边分别加0、1、2、3个保护碱基后的切断情况。
结果显示,基本上所有限制酶,在其酶切位点旁边加上3个以上的保护碱基后,可以对其酶切位点进行有效切断。
一般来讲,在酶切位点前加入两个GC碱基,因为如果保护碱基为AT的话,保护碱基在PCR产物的末端,AT之间只有两个氢键,结合力差,容易在末端产生单链,这样的话限制性内切酶就无法作用。
其实加保护碱基的多少,是具体情况具体讨论,比如HindIII、BamHI等就得有三个保护碱基。
少了一个就无法切动。
保护碱基的作用限制性内切酶识别特定的DNA序列,除此之外,酶蛋白还要占据识别位点两边的若干个碱基,这些碱基对内切酶稳定的结合到DNA双链并发挥切割DNA作用是有很大影响的,被称为保护碱基。
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的,见附表。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
酶切位点保护碱基表6PCR引物设计原则信息来源:本站原创更新时间:2004-12-21 0:44:00PCR引物设计的目的是为了找到一对合适的核苷酸片段,使其能有效地扩增模板DNA序列。
因此,引物的优劣直接关系到PCR的特异性与成功与否。
要设计引物首先要找到DNA序列的保守区。
同时应预测将要扩增的片段单链是否形成二级结构。
如这个区域单链能形成二级结构,就要避开它。
如这一段不能形成二级结构,那就可以在这一区域设计引物。
现在可以在这一保守区域里设计一对引物。
一般引物长度为15~30碱基,扩增片段长度为100~600碱基对。
让我们先看看P1引物。
一般引物序列中G+C含量一般为40%~60%。
而且四种碱基的分布最好随机。
不要有聚嘌呤或聚嘧啶存在。
否则P1引物设计的就不合理。
应重新寻找区域设计引物。
同时引物之间也不能有互补性,一般一对引物间不应多于4个连续碱基的互补。
引物确定以后,可以对引物进行必要的修饰,例如可以在引物的5′端加酶切位点序列;标记生物素、荧光素、地高辛等,这对扩增的特异性影响不大。
但3′端绝对不能进行任何修饰,因为引物的延伸是从3′端开始的。
这里还需提醒的是3′端不要终止于密码子的第3位,因为密码子第3位易发生简并,会影响扩增的特异性与效率。
综上所述我们可以归纳十条PCR引物的设计原则:①引物应用核酸系列保守区设计并具有特异性。
②产物不能形成二级结构。
③引物长度一般在15~30碱基之间。
④G+C含量在40%~60%之间。
⑤碱基要随机分布。
⑥引物自身不能有连续4个碱基的互补。
⑦引物之间不能有连续4个碱基的互补。
⑧引物5′端可以修饰。
⑨引物3′端不可修饰。
⑩引物3′端要避开密码子的第3位。
PCR引物设计的目的是找到一对合适的核苷酸片段,使其能有效地扩增模板DNA序列。
如前述,引物的优劣直接关系到PCR的特异性与成功与否。
对引物的设计不可能有一种包罗万象的规则确保PCR的成功,但遵循某些原则,则有助于引物的设计。