铜催化点击化学反应综述
- 格式:pdf
- 大小:268.90 KB
- 文档页数:4

《铜基催化剂的制备及其电化学还原CO2制乙烯的研究》篇一一、引言随着全球能源需求的增长和环境污染问题的加剧,如何有效利用和转化二氧化碳(CO2)已成为科学界和工业界的重要研究课题。
电化学还原CO2技术作为一种新兴的二氧化碳利用技术,具有高选择性、高效率、环境友好等优点,而铜基催化剂作为其关键组成部分,对CO2的转化和还原起着至关重要的作用。
本文将详细介绍铜基催化剂的制备方法,以及其在电化学还原CO2制乙烯的研究进展。
二、铜基催化剂的制备1. 材料选择与预处理制备铜基催化剂的材料主要选用高纯度的铜源,如硝酸铜、氧化铜等。
在制备前,需对原料进行适当的预处理,如烘干、研磨等,以提高其反应活性。
2. 催化剂制备方法(1)溶胶凝胶法:将铜源溶解在适当的溶剂中,加入适量的表面活性剂或稳定剂,通过控制反应条件,形成稳定的溶胶凝胶体系,再经过热处理得到铜基催化剂。
(2)共沉淀法:将铜源与其它金属盐溶液混合,通过调节pH值等条件,使金属离子共沉淀形成复合物,再经过热处理得到铜基催化剂。
(3)化学气相沉积法:通过将含铜化合物的前驱体在高温下气化,并在基底上沉积形成铜基催化剂。
三、电化学还原CO2制乙烯的研究1. 反应原理电化学还原CO2制乙烯的反应过程中,铜基催化剂作为阴极材料,通过施加一定的电压和电流,使CO2在催化剂表面发生还原反应,生成乙烯等有机物。
2. 实验方法与步骤(1)电解液的选择:选择适当的电解液对反应过程具有重要影响。
通常选择离子导电性好、电化学窗口宽的溶液作为电解液。
(2)实验装置的搭建:搭建电化学工作站和电解池等实验装置,将铜基催化剂作为阴极材料置于电解池中。
(3)实验操作:在一定的电压和电流条件下进行电解反应,记录反应过程中的电流、电压、产气量等数据。
通过调整实验参数和优化反应条件,提高乙烯的产量和选择性。
3. 结果与讨论通过对不同条件下得到的实验数据进行分析比较,可以得出以下结论:在一定的电压和电流条件下,铜基催化剂具有较高的电化学还原CO2制乙烯的活性和选择性。

几种温和铜催化反应简介在过去的几年当中,对于碳杂键的建立,铜催化剂被认为是一种有效的物质,并且在工业生产中得到了使用。
在这一类型的反应中,许多的温和的不同于以往在反应中使用的有害的有机溶剂被开发。
1 室温铜催化芳基硼酸与氨水偶联合成一级芳胺一级芳胺的经典合成方法就是在高温,高压和封闭容器中采用钯催化卤代芳烃与氨气或氨气的替代物如Li[N(SiMe3)2],Zn[N (SiMe3)2]反应,硝基化合物的还原,羧酸的Curtis 重排,Buchwald 反应等方法来制备。
本文介绍一种新的合成一级芳胺的新方法,即在室温和常压下,使用Cu2O 作催化剂,芳基硼酸与氨水在甲醇溶液中反应合成了一级芳胺。
反应容器不需要密闭,不需要惰性气体保护,也不需要在反应体系中加入任何配体或添加剂。
以下是一些典型硼酸的反应收率:从表中可以醛,酯,卤代物等官能团都能够不受影响,反应条件十分温和,可操作性强。
2 一种在温和铜催化条件下合成喹唑啉酮的有效方法在CuI 催化下,利用邻卤代苯甲酸衍生物与脒或胍一锅反应即可制备喹唑啉酮衍生物。
这种方法反应条件温和,反应体系中不需要加入任何配体和添加剂。
卤代苯甲酸衍生物的活性顺序为:碘代物>溴代物>氯代物。
3 铜催化室温条件下直接将芳基甲烷转化为芳基腈芳基腈主要是通过芳香胺的Sandmeyer 反应,在过渡金属催化下芳香卤代物与氰化物的反应,酰胺的脱水反应来制备。
本文介绍直接将芳基甲烷转化为芳基腈的方法。
该方法简单,高效,不需要高温,高压以及贵重金属催化,并且成功地运用于具有抗小核糖核酸病毒活性分子的合成中。
进一步的研究表明芳基甲烷中如果连有供电子基团,对反应有利,并且在反应过程中酯,醚等官能团以及Boc 和MOM 保护基都能不受影响。
4 铜盐催化下杂环与芳基溴的直接C-芳基化反应杂环芳基化反应通常是在Rh,Ru 等贵重金属催化下完成的。
Daugulis 报道了铜催化的杂环芳基化反应,但是反应主要针对活性高的碘代物有效,并且有叔丁基芳基醚副产物生成。

单原子cu电催化综述单原子Cu电催化综述引言:电催化技术是一种将电能转化为化学能的方法,广泛应用于能源转换、环境保护和有机合成等领域。
近年来,单原子催化剂作为一种新兴的电催化剂材料,引起了广泛的关注和研究。
本综述将以单原子Cu电催化为主题,对其在电催化领域的应用和研究进展进行综述。
一、单原子Cu电催化的优势单原子Cu电催化剂具有以下几个优势:首先,单原子Cu催化剂具有高比表面积和丰富的活性位点,能够提供更好的催化活性;其次,单原子Cu催化剂具有良好的稳定性,能够长时间保持高效的催化活性;最后,单原子Cu催化剂还具有可调控性强的特点,可以通过调节制备方法和表面修饰等手段,实现对催化性能的优化。
二、单原子Cu电催化的制备方法单原子Cu电催化剂的制备方法主要包括化学还原法、模板法、原位合成法和表面修饰法等。
其中,化学还原法是制备单原子Cu催化剂的常用方法,通过选择适当的还原剂和溶剂,可以在合适的反应条件下得到单原子Cu催化剂。
模板法则是利用模板分子或材料作为载体,将Cu原子分散在载体上,然后去除载体获得单原子Cu催化剂。
原位合成法是在催化剂合成过程中,在合成体系中加入一些添加剂,通过调节添加剂浓度和反应条件等,可以得到单原子Cu 催化剂。
表面修饰法是在已有的催化剂表面进行修饰,通过改变表面的化学组成和结构,实现对催化性能的调控。
三、单原子Cu电催化的应用领域单原子Cu催化剂在电催化领域有广泛的应用。
首先,单原子Cu催化剂在电催化水分解产氢反应中表现出优异的催化性能。
研究表明,单原子Cu催化剂具有较低的能量损失和较高的稳定性,能够显著提高水分解产氢反应的效率。
其次,单原子Cu催化剂还可以应用于电催化CO2还原反应,实现CO2的高效转化。
研究表明,单原子Cu催化剂具有较高的CO2吸附能力和较低的CO2还原活化能,能够有效促进CO2的还原反应。
此外,单原子Cu催化剂还可以应用于电催化氧还原反应、电催化有机合成等领域,展现出良好的催化性能。

铜系催化剂应用综述医药化工学院化学工程与工艺专业学生:陈立峰陈峰舒文强陈灵指导老师:摘要铜作为催化剂, 具有价格低廉、毒性低等优点, 此外, Cu物种比较温和而且配体简单, 正因为如此, 应用Cu 盐进行催化化学反应是目前非常热门的一个领域。
以下介绍Cu催化剂应用的研究与新应用。
关键词铜系催化剂合成甲醇催化剂铜系催化剂热分析铜系催化剂热相分析1 铜系催化剂的各方面应用2.1 Cu 催化交叉偶联反应2.1.1 Ullmann 反应早期的Ullmann 反应局限于卤代芳烃和芳基亲核化合物( 如芳胺、酚类、硫酚类等) 之间的偶联. 尽管实际起作用的是一价铜络合物, 在反应中人们通常使用过量的铜粉. 反应的温度通常高达200℃, 反应的后续处理困难, 反应产物复杂, 反应的产率也不高. 尽管如此, 由于在早期人们没有其它办法来实现亲电性sp2 碳与亲核试剂之间的直接偶联,Ullmann 反应仍然被合成工作者大量使用. 1998 年, 马大为等报道了卤代芳烃与A-氨基酸之间进行偶联得到N-芳基-A-氨基酸的反应. 这一反应使用CuI作催化剂, 溶剂为DMA, 反应条件较为温和. 利用该反应, 他们合成了重要的医药试剂Benzolactam-V8.2001 年, 马大为等又将上述催化体系应用到B-氨基酸的芳基化中, 同样取得了很好的结果(Eq. 1) . 他们发现B-氨基酸也可以加速反应的进行, 其机理类似于A- 氨基酸的芳基化过程. 利用这一反应, 他们成功地合成了SB-214857.(1) Buchwald 研究组最终找到了一种通用、温和、简单, 而且高效的碳、氮偶联方法. 使用该方法, Buchwald 等高产率地合成了一系列的芳香胺、脂肪胺、酰胺以及吲哚等芳基化产物. 作为一个成功的例子, 下面的成环反应可以使用CuI 作为催化剂, N, Nc-二甲基乙二胺作为辅助配体, 通过分子内的胺芳基化来实现( Eq. 2) . 该反应可在室温下进行, 产率很高.(2)同时, 他们还发现该催化体系有很好的选择性. 在单取代酰基肼的氮芳基化中, 以叔丁氧甲酰肼为底物和间位和对位取代的碘苯进行的反应时, 只是得到N-芳基化合物A,而苯甲酰肼和邻位取代的碘苯进行反应时, 得到的是Nc-芳基化合物B ( Eq. 3)(3) Buchwald 等最近将这一催化体系应用到碳、卤偶联化合物的制备. 他们发现以下的反应可以高效地将芳烃或者烯烃的溴化物转化为碘化物( Eq. 4 .(4)Cuny 等使用( CuOTf) 2PhMe 作为催化剂制备了具有生物活性的2-羟基-2c甲氧基二苯基醚(Eq.5) . 他们还应用该反应简捷地合成了有助于神经生长的药物verbenachalcone.(5)Venkataraman[ 46] 报道了CuI 催化的碳-硒交叉偶联反应( Eq. 6) . 该反应使用CuI 和2, 2c- 联喹啉亚铜作为催化体系, 以叔丁基钠( 对于富电子的芳香碘) 和碳酸钾( 对于贫电子的芳香碘) 作为碱, 合成了十八种的碳) 硒化合物, 最高的产率达到92%.(6)2.1.2Stille 反应Stille 反应通常是由钯催化的芳基锡化合物与芳基卤代物之间的交叉偶联反应. 目前该反应已经广泛地被应用在有机合成中, 用于制备各种不对称的芳香交叉偶联产物.由于锡烷化合物对于水汽和空气都是稳定的, 并且对很多的官能团表现出化学惰性, 因而它们应用范围很广. 同时, 由于Stille 反应中生成不溶的锡盐类, 所以可以很容易实现目标产物与副产物的分离.尽管Stille 反应通常由Pd 来催化, Roth 等。

铜催化N-芳基化反应药物合成专业毕业论文铜催化N-芳基化反应是一种重要的有机合成方法,可以用于合成多种具有药物活性的化合物。
本文将从铜催化N-芳基化反应的原理、机理和影响因素等方面进行阐述,并以药物合成为应用场景,探讨其在药物合成中的应用。
一、铜催化N-芳基化反应的原理与机理铜催化N-芳基化反应是一种通过将亲电芳香化合物加到含有N-叔丁基苯磺酰氨基的底物上,使其发生亲核反应,进而形成N-芳基化合物的有机转化反应。
其反应机理如下:首先,底物通过铜催化发生亲核取代反应,失去叔丁基空间位阻,使底物的N原子更容易进行亲电芳香化反应。
然后,亲电芳香化合物进一步发生亲核反应,与底物N原子形成氮上的碳-氮化合物。
最后,通过酸解离或碱解离去除N-磺酰基,得到N-芳基化合物。
反应中铜离子通过将底物上的N-叔丁基苯磺酰氨基进行解离,使其失去位阻,提高其亲核反应的效率和底物的反应性。
同时,铜离子还可以使亲电芳香化合物更容易发生亲核反应,形成氮上的碳-氮化合物。
二、影响铜催化N-芳基化反应的因素在铜催化N-芳基化反应中,反应条件是影响反应效果的重要因素。
反应条件包括反应温度、反应时间、底物浓度、亲电芳香化合物的种类和含量、催化剂种类和含量等。
1.反应温度:反应温度是影响反应速率和产物选择性的重要因素。
通常情况下,反应温度在60-120°C之间,较高的反应温度可以促进反应速率,但同时也会影响产物选择性。
2.反应时间:反应时间对反应速率和产物选择性也有很大影响。
在反应前期,反应中可能会产生一些不利于产物选择性的中间体,但随着反应时间的延长,这些中间体会继续反应,最终形成稳定产物。
3.底物浓度:底物浓度对反应速率和产物选择性都有影响。
较低的底物浓度可以提高产物选择性,但过低的底物浓度会降低反应速率。
4.亲电芳香化合物种类和含量:典型的亲电芳香化合物包括硝基苯和卤代苯等。
不同亲电芳香化合物的种类和含量对反应速率和选择性也有明显影响。

buchwald铜催化反应
Buchwald铜催化反应是一种重要的有机合成方法,它是以铜作为催化剂,在有机合成中实现碳-氮键或碳-碳键的形成。
该反应以其高效、高选择性和广泛适用性而受到广泛关注。
Buchwald铜催化反应最常见的应用是在C-N键的构建中,即通过催化剂促使芳香胺和芳香卤化物之间形成新的C-N键。
这种反应对于药物合成、材料科学以及天然产物合成等领域具有重要意义。
在Buchwald铜催化反应中,通常使用的催化剂是含有铜和配体的配合物。
这些配合物能够稳定铜离子,并调节反应的速率和选择性。
常用的配体有JohnPhos、XPhos、BINAP等。
这些配体能够与铜形成配位键,并提供空间位阻,从而控制反应的立体化学和催化活性。
Buchwald铜催化反应的反应条件相对温和,通常在室温或略高温下进行。
反应溶剂可以是常见的有机溶剂,如二甲基甲酰胺(DMF)、二氯甲烷(DCM)等。
反应时间一般较短,通常在几小时到一天之间。
Buchwald铜催化反应的机理还在研究中,但一般认为其机理涉
及铜催化剂的活化、配体的配位和底物的反应等步骤。
具体的机理
细节可能因反应底物和配体的不同而有所差异。
总的来说,Buchwald铜催化反应是一种重要的有机合成方法,
通过铜催化剂和配体的协同作用,实现了碳-氮键或碳-碳键的形成。
该反应在有机合成中具有广泛的应用前景,并为合成化学领域的研
究和发展提供了重要的工具和方法。
毕业论文文献综述应用化学铜催化剂的新应用与发展1、引言1.1铜的简介铜是一种传统而又现代重要的金属材料。
在人类使用的所有材料中,铜对人类文明的影响最为显著。
从人类文明初期直至如今,铜对社会的不断进步做出了重大贡献。
随着人类社会向电气化、自动化、信息化和网络化的方向迈进,铜在生产建设、人民生活以及高新技术上的重要作用日益明显。
当前微电子工业中“铜芯片”革命的兴起,以及采用ADSL(数字用户专线)技术使标准铜电话线同时运载高速数据得以实现,就很好的证明了,铜不仅是一种传统的非常有用的金属,而且还是重要的现代高新技术材料。
铜的元素符号为Cu,化学元素周期表第四周期IB族元素,原子序数29,原子量63.54,电子构型8-18-1(3d104s1);比重8.92g/cm3,熔点1083℃,沸点2360℃。
纯铜呈浅玫瑰色或淡红色,表面形成氧化铜膜后,外观呈紫铜色。
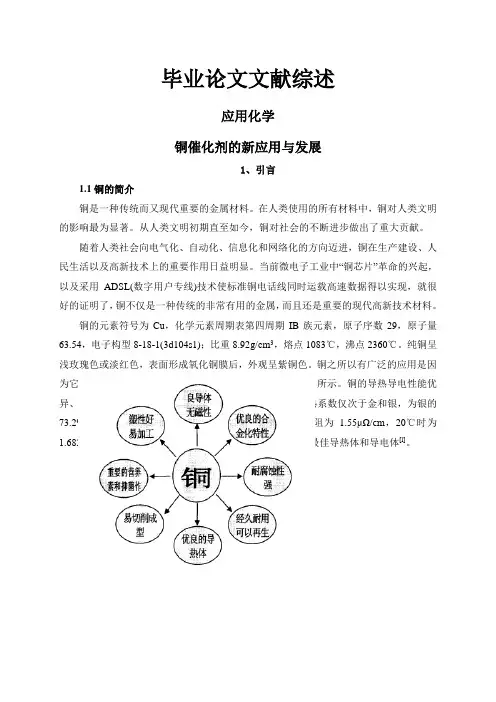
铜之所以有广泛的应用是因为它具有许多优异的物理和化学性能,其简要特性如图1.1所示。
铜的导热导电性能优异、塑性好、易加工、耐腐蚀、无磁性、美观耐用。
其导热系数仅次于金和银,为银的73.2%,金的88.8%;作为电的良导体,O℃时铜的比电阻为 1.55μΩ/cm,20℃时为1.682μΩ/cm,由于金和银价格昂贵,因而铜是广泛应用的最佳导热体和导电体[l]。
图1.1铜的简要特性示意图铜还能与许多金属形成合金。
例如,黄铜为铜与锌的合金,白铜为铜与镍的合金,青铜为铜与铝、锡等元素形成的合金。
铜中加入合金元素,可以进一步提高其强度、硬度、弹性、易切削性、耐磨性以及抗腐蚀等方面的性能,以满足不同的使用要求。
1.2铜的应用[2]铜与人类关系非常密切,被广泛地应用于电气、轻工、机械制造、建筑工业、国防工业等领域,在我国有色金属材料的消费中仅次于铝。
铜在我国的大致的消费结构如图1.2所示。
铜在电气、电子工业中应用最广,用量最大,占总消费量一半以上。
具体用于各种电缆和导线,电机和变压器的绕阻,开关以及印刷线路板等。

生物正交化学中的点击化学反应:探索点击化学反应在活细胞标记与药物靶向递送中的应用摘要点击化学反应作为生物正交化学的重要工具,因其高度选择性、快速反应和生物相容性,在活细胞标记和药物靶向递送领域展现出巨大潜力。
本文深入探讨了点击化学反应的原理、类型、特点以及在上述领域的应用。
通过分析点击化学反应的优势、挑战和未来发展方向,本文旨在展示其在生命科学研究和医学应用中的重要价值。
引言生物正交化学(Bioorthogonal Chemistry)是指一类能够在活体系统中进行,而不干扰生物体内正常生化过程的化学反应。
点击化学(Click Chemistry)作为生物正交化学的重要组成部分,具有高度选择性、反应条件温和、产率高、副产物少等优点,被广泛应用于活细胞标记、生物分子成像、药物靶向递送等领域。
点击化学反应的原理与类型点击化学反应通常涉及两种相互正交的反应基团,它们在生理条件下能够快速、高效地发生反应,生成稳定的产物。
常见的点击化学反应类型包括:1. 铜催化的叠氮-炔烃环加成反应(CuAAC):该反应利用铜离子作为催化剂,将叠氮基团(-N3)和炔烃基团(-C≡CH)连接起来,生成稳定的三唑环。
2. 无铜催化的叠氮-炔烃环加成反应(SPAAC):该反应利用环状炔烃的环张力,无需铜离子催化即可与叠氮基团发生反应,生成三唑环。
3. 应变促进的炔烃-叠氮化物环加成反应(SPANC):该反应利用环辛炔的环张力,无需铜离子催化即可与叠氮基团发生反应,生成三唑环。
4. 四嗪-反式环辛烯环加成反应(TCO-Tetrazine Ligation):该反应利用四嗪和反式环辛烯之间的Diels-Alder反应,生成稳定的环加成产物。
点击化学反应在活细胞标记中的应用点击化学反应在活细胞标记中具有以下优势:1. 高特异性:点击化学反应的反应基团具有高度选择性,只与特定的反应伙伴发生反应,避免了对细胞内其他生物分子的干扰。
2. 快速反应:点击化学反应通常在温和条件下快速进行,可以在短时间内完成标记过程。

点击化学及其在高分子研究中的应用摘要点击化学是一种运用高产率、高选择性的高效化学方法来快速合成化合物的模块化合成手段。
该方法具备产量高、效率高、副反应少、反应条件温和、分离提纯简单、环境污小等优点,因此得到了广泛的应用。
目前点击化学发展极为迅速,涉及到了各个领域,特别是在功能聚合物、大分子化合物、表面修饰、药物开发、生物与化学传感器等方面取得了瞩目的成就。
本文论述了点击化学反应的基本概念、反应类型和反应特征,重点介绍了点击化学在功能聚合物、大分子化合物及表面修饰方面的应用。
关键词:点击化学;功能聚合物;大分子化合物;表面修饰1、引言材料是人类生活和生产的基础,从人类发展的历史来看,每一种重要材料的发现和利用都给社会生产力和人类生活带来巨大变化。
随着科技的发展,材料科学与生物科学、生物工程、化学、物理、信息科学和环境科学等不断交叉渗透,促进了新材料的发明和利用,但仍不能满足人们对材料功能日益增长的需求。
因此,迫切需要新技术、新方法为新材料的研发注入新的活力。
“Click Chemistry”又称点击化学,是一种快速合成大量化合物的新方法,在众多领域得到了迅速的发展,如DNA[1]、自组装[2-3]、表面修饰[4-5]、超分子化学[6-7]、树枝状分子[8-9]、功能聚合物[10]、生物及化学传感器[11-14]和生物医药领域[15-16]等方面展示广发的应用前景。
2、点击化学简述点击化学是由2001年诺贝尔化学奖得主美国化学家Sharpless首次提出的[17],他希望化学反应像操作个人电脑一样(仅需点击鼠标)可控、简单、高效、快捷。
点击化学是一种快速合成一系列含有C-X-C原子键接单元化合物的组合化学新方法,与传统的化学合成技术相比,点击化学更能适应分子多样性的要求,用少量简单可靠和高选择性的化学转变来获得更广泛的分子多样性结构。
点击化学是指具有以下特征的一类化学反应:(1)反应模块化,主要是Cu+催化条件下的叠氮化物和端位炔Husigen 1,3-偶极环加成反应(CuAAC);(2)使用范围广,如有机合成、蛋白质和DNA分子标记等;(3)产率高,副产物少,产物通过简单的结晶或蒸馏即可分离纯化;(4)较好的立体选择性;(5)反应条件简单,产物对水和氧气都不敏感;(6)原料和反应试剂易得;(7)反应不使用溶剂或使用水等良性溶剂;(8)产物在生理条件下稳定,有较好的生物相容性;(9)反应需要较高的热力学驱动力(>84kJ/mol);(10)符合原子经济性。

一价铜催化的三键-叠氮点击环化反应的机理和应用[原文] Bock, V. D.; Hiemstra, H.; van Maarseveen, J. H. Eur, J. Org. Chem.2006, 51-68.摘要在一价铜催化下,三键-叠氮环化反应生成1, 4-二取代的1, 2, 3-三唑,这个反应具有高效性和广谱适用性,因此被称为点击化学(Click Chemistry)。
在本文中我们从反应的机理出发对大量实验结果进行综合分析(包括液相和固相),主要集中在催化剂的生成,溶剂,底物效应以及碱和配体的选择配对。
关键字:点击化学; 成环反应; 杂环; 分子多样性; 叠氮化合物.1.一价铜催化的三键叠氮成环反应简介随着人类对新的化学材料和生物活性分子需求的日益增长,化学家们对具有新颖结构的活性化合物的开发已经不能满足要求了[1]。
点击化学(一类高选择性高效形成杂环连接单元的反应)的出现,为解决此问题提供了转机[2]。
一个反应能被称为点击反应必须具备以下条件:温和的反应条件,简单的操作分离过程,并且可以通过一些反应模板能高效地创建分子库[2]。
利用点击化学可以从现有的分子可靠高效地创建新的活性化合物库。
Sharpless小组已经找到了许多能满足点击化学标准的反应[2],其中一价铜催化的三键和叠氮1, 3偶极成环[3]生成1, 2, 3-三唑的Huisgen反应被证明是到目前为止最有效的反应。
在有机合成中,三键和叠氮是两种常见的官能团,能够很方便地被引入到底物中并且这两种官能团对氧气和水不敏感,能够经受众多的有机反应条件[4, 5],对具有多官能团的生物分子也表现出了一定的惰性。
在大多数的情况下,这两个官能团能够在合适的条件下被引入到目标分子中并且在接下来的一系列转化过程中能保持一定的惰性[6]。
尽管叠氮官能团在热力学上是一个不稳定的官能团,它容易分解,但由于反应动力学因素使得脂肪族的叠氮化合物稳定存在直到遇到亲偶极体发生偶极加成反应[5],也正是因为三键和叠氮官能团的动力学稳定性,在没有催化剂存在下,两者之间的成环反应非常慢,通常需要加热或是延长反应时间才能完成[7, 8]。

铜催化剂参与反应全文共四篇示例,供读者参考第一篇示例:铜催化剂是一种常用的催化剂,它在许多化学反应中发挥着重要作用。
铜催化剂能够促进反应的进行,降低反应的能量,加速反应速率,并提高反应的选择性。
铜催化剂在有机合成、氧化还原反应、氨基化反应等领域中得到了广泛应用。
铜催化剂参与的反应有很多种,下面我们就来详细介绍一些常见的反应类型及其机理。
1. 烯烃的环化反应铜催化剂在烯烃的环化反应中发挥重要作用。
烯烃经过环化反应后可以形成环烷烃或环芳烃等化合物,这对于有机合成有着重要意义。
铜催化剂通常能够催化烯烃的氧化环化反应,将烯烃转化为环化合物。
这种反应通常采用氧化剂作为辅助剂,如过氧酸、金属叶酸等。
2. 羧基的偶联反应在有机合成领域,羧基的偶联反应也是一种常见的反应类型。
铜催化剂可以促进羧基之间的偶联反应,形成碳-碳键或碳-氧键。
这种反应通常以铜催化剂为主导,通过活化羧基中的碳原子或氧原子来实现羧基之间的偶联。
铜催化剂还常常用于炔烃的环加成反应中。
炔烃经过环加成反应后可以形成环戊烷或环戊二烯等化合物,这对于有机合成也有着极大的意义。
铜催化剂通常能够加速炔烃的环加成反应,降低反应的活化能垒,提高反应的速率。
4. 芳香族化合物的取代反应5. 碳碳键形成反应铜催化剂在碳碳键形成反应中也是一个重要的催化剂。
碳碳键的形成对于有机合成来说是至关重要的,而铜催化剂可以促进碳碳键的形成反应,降低反应的活化能,提高反应的速率,从而实现碳碳键的高效合成。
第二篇示例:铜催化剂是一种在许多有机合成反应中起着重要作用的催化剂。
铜催化剂具有良好的催化活性和选择性,广泛应用于碳-碳键和碳-氮键的形成以及氢原子转移等反应中。
本文将重点介绍铜催化剂在有机合成中的应用和作用原理。
我们来介绍一下铜催化剂在碳-碳键和碳-氮键形成反应中的应用。
在有机合成中,碳-碳键的形成对于构建有机分子结构至关重要。
铜催化的Cross-Coupling反应是一种常用的方法来实现碳-碳键的形成。
铜催化剂是一种常用的催化剂,可在多种有机合成反应中发挥重要作用。
以下是一些常见的铜催化反应类型:
偶联反应:铜催化剂可以促进芳香和脂肪基化合物之间的偶联反应。
常见的偶联反应包括Ullmann偶联反应、Sonogashira偶联反应、Buchwald-Hartwig偶联反应等。
环化反应:铜催化剂在环化反应中可以促进C-C键或C-N键的形成。
例如,铜催化的氧化亲和反应可以将不饱和化合物转化为环状化合物。
氧化反应:铜催化剂可以参与氧化反应中的氧化还原过程。
一种常见的铜催化氧化反应是铜催化的氨基醇氧化反应。
羟基化反应:铜催化剂可以催化芳香化合物或烯烃与水或醇发生羟基化反应。
例如,铜催化的某些叠氮化合物和芳基硼酸的反应可以得到醇类产物。
环加成反应:铜催化剂可以促进烯烃和另一个分子(如亚胺、亚硫酰胺等)之间的环加成反应,形成环状化合物。
需要注意的是,具体的反应机理和条件会因具体的反应类型而有所不同。
铜催化剂的选择、反应底物的配体修饰以及反应条件的优化都会对反应的效果产生影响。
因此,在使用铜催化剂参与反应时,需要根据具体反应的要求进行设计和优化,以获得理想的产物收率和选择性。
点击化学在生物医用高分子中的应用一、本文概述点击化学,作为一种高效、精确的合成方法,近年来在化学领域引起了广泛关注。
其独特的反应特性,如反应速度快、产物纯度高、副反应少等,使得点击化学在材料科学、生物医学等多个领域都有着广泛的应用前景。
本文将重点探讨点击化学在生物医用高分子领域的应用,分析其在该领域的发展现状、优势及挑战,并展望未来的发展趋势。
在生物医用高分子领域,点击化学的应用主要集中在高分子材料的合成、改性和生物活性分子的偶联等方面。
通过点击化学反应,可以实现对高分子链结构的精确调控,从而制备出具有特定功能和生物活性的高分子材料。
这些材料在药物载体、组织工程、生物传感器等领域具有广泛的应用价值。
本文将首先介绍点击化学的基本原理和常用方法,然后重点分析点击化学在生物医用高分子合成和改性中的应用案例,探讨其在实际应用中的优势和局限性。
还将讨论点击化学在生物活性分子偶联、药物递送系统以及生物医学成像等方面的应用,并展望未来的研究方向和应用前景。
通过本文的阐述,旨在为读者提供一个全面、深入的了解点击化学在生物医用高分子领域应用的视角,为推动该领域的发展提供参考和借鉴。
二、点击化学基本原理点击化学,又称“动态共价化学”,是由夏普莱斯教授在2001年首次提出的一种合成概念。
其核心在于利用高选择性的化学反应,通过简单的操作,快速、高效地完成分子间的连接。
点击化学的核心理念在于“简单、快速、高效、选择性好”,这一理念在化学合成领域引起了极大的反响。
点击化学的基本原理主要基于几种具有高反应活性的官能团之间的反应,如叠氮-炔烃的1,3-偶极环加成反应(Huisgen1,3-dipolar cycloaddition)、巯基-烯/炔的点击反应(Thiol-ene/Thiol-yne reactions)、Diels-Alder反应、氮杂-Diels-Alder反应等。
这些反应通常具有高度的选择性,可以在温和的条件下快速进行,而且不需要严格的反应条件控制,如无水无氧等。
铜基电催化方法综述-回复什么是铜基电催化?为什么铜被广泛应用于电催化反应中?铜基电催化是指利用铜作为电子转移媒介,在电化学反应中加速化学反应进程的一种方法。
近年来,铜基电催化已成为有机合成和能源转化领域中备受关注的研究方向。
铜作为一种常见而且廉价的金属,具有较高的电导率和电子转移速率,因此被广泛应用于电催化反应中。
对于铜基电催化方法的综述,可以从以下几个方面进行探讨:1. 铜基电催化的基本原理和机制:铜基电催化的基本原理是利用电子传递过程中铜的氧化还原性质,促使反应底物的氧化还原过程。
电子传递过程可通过阴极和阳极上的电荷传动实现,从而加速化学反应的进行。
铜基电催化涉及多种电催化反应机制,如电化学还原、电化学氧化、电子转移和催化循环等。
2. 铜基电催化在有机合成中的应用:铜基电催化在有机合成中具有独特的优势,例如它可以在温和的条件下实现选择性催化,催化剂寿命较长,反应底物范围广等。
近年来,铜基电催化已被应用于多种有机反应中,例如偶联反应、羰基化反应、烯炔化反应等。
其中,偶联反应是铜基电催化的典型应用之一,如Suzuki反应、Sonogashira反应、Goldberg反应等。
3. 铜基电催化在能源转化中的应用:铜基电催化也在能源转化领域中展示出巨大的潜力。
例如,在电解水制氢过程中,使用铜作为电极催化剂可以提高氢气产率和电化学性能。
此外,铜基电催化还可以应用于二氧化碳还原、氧气还原以及锂离子电池等能源转化过程中。
4. 铜基电催化的挑战与展望:尽管铜基电催化在有机合成和能源转化领域中显示出了巨大的潜力,但仍然面临一些挑战。
其中之一是提高反应的选择性和效率。
此外,铜基电催化的机理和反应条件还需要进一步研究和探索。
未来的发展方向可能包括设计和合成新型的铜基催化剂,探索新的反应机制,以及改进电催化反应的条件等。
综上所述,铜基电催化作为一种有效的电催化方法,已得到广泛应用于有机合成和能源转化领域。
随着对铜基电催化的研究深入进行,相信将会有更多的新催化剂和反应机制被发现和应用,进一步推动电催化领域的发展。
点击化学反应在生物标记物中的应用
点击化学反应(Click Chemistry)是一种高效的化学合成方法,应用广泛且效
果显著。
在生物标记物中,点击化学反应被广泛应用于检测、诊断和治疗疾病,为生命科学领域带来了革命性变革。
点击化学反应的原理简单明了,反应条件温和,产率高。
这使得点击化学反应
成为生物标记物研究中的重要工具。
其中,最为常见的点击化学反应之一就是铜催化的炔基-叠氮环加成反应,具有高度选择性和底物范围广泛等优点,因此被广泛
应用于药物合成和生物标记物制备中。
在生物标记物领域,点击化学反应被用于构建多功能的生物标记物。
通过引入
点击反应位点,可以实现生物标记物的快速合成和修饰,从而实现对生物分子的高效检测和定位。
例如,科学家们利用点击化学反应成功制备了一系列具有特异性的荧光标记物,用于细胞成像和蛋白质检测等研究。
此外,点击化学反应也被广泛应用于药物传递系统的设计和制备中。
通过点击
化学反应引入靶向基团和药物分子,可以实现药物的靶向传递和释放,提高药物的治疗效果并减少副作用。
这为肿瘤治疗等领域带来了新的希望,为个性化医疗提供了新的途径。
总的来说,点击化学反应在生物标记物中的应用具有广泛的前景和重要的意义。
随着技术的不断进步和创新,点击化学反应在生物标记物研究中将发挥越来越重要的作用,推动生命科学领域的发展和进步。
希望未来能够有更多的研究者投入到这一领域,共同发掘点击化学反应的更多潜力,为人类健康和生命科学的发展作出更大的贡献。
铜作催化剂(范文3篇)以下是网友分享的关于铜作催化剂的资料3篇,希望对您有所帮助,就爱阅读感谢您的支持。
《铜作催化剂范文一》一.概述铜基合成甲醇催化剂须经还原后才具有活性。
还原反应是一个强放热反应,反应式如下所示:CuO + H2 ==== Cu + H2O + 86.7KJ/mol因此,在还原过程中应特别注意控制催化剂床层温度,防止催化剂过热发生铜晶粒烧结而损害催化剂活性。
还原操作是开车过程中很重要的一个操作环节。
每炉催化剂活性的高低,除与催化剂自身的生产质量和装填质量有关外,很大程度上还取决于催化剂还原质量的好坏,它将对装置的生产能力产生长远的影响。
因此,必须严格、细致、认真地进行还原操作。
XNC-98型合成甲醇催化剂采用低氢(1%H2)还原工艺。
催化剂在还原过程中出水量约为催化剂重量的18×10-2~20×10-2,其中物理水3×10-2~5×10-2,化学水13×10-2~15×10-2。
如果还原气中含CO,则生成的水少些。
二.XNC-98型低压甲醇合成催化剂的组成、物性和技术指标1.外观颜色及形状:黑色有金属光泽的圆柱体2.外形尺寸,mm Φ5×(4.5~5.0)3.堆密度,kg/L :1.35~1.45 4.侧压抗破碎强度,N/cm :≥200 5.化学组成,(×10-2 m/m):6.催化剂活性在本催化剂质量检验标准规定的活性检测条件下催化剂活性为:230℃时,催化剂的时空收率≥ 1.20kg/L h;250℃时,催化剂的时空收率≥ 1.55kg/L h。
7.催化剂使用寿命在正常条件下运行寿命为2年以上。
三.还原前的准备工作1.催化剂装填完毕后,应用清洁的空气(或氮气)将催化剂粉末从合成塔中吹除干净。
2.公用工程准备就绪。
3.循环气压缩机、合成气压缩机均已调试合格。
4.合成系统气密性试验合格。
5.合成系统的电器、仪器、仪表已调试合格,仪表已校准(合成塔进出口温度、压力及合成回路中各流量显示仪表必须严格校准)。
铜基电催化二氧化碳还原综述
铜基电催化二氧化碳还原是一种利用电能将二氧化碳转化为有价值的碳氢化合物的方法,近年来受到了广泛关注。
作为一种高效、环保的转化技术,它被认为是解决全球气候变化和碳排放问题的重要途径之一。
铜基电催化二氧化碳还原的原理是基于电化学反应,通过电解液中的铜基催化剂的作用,将二氧化碳还原为碳氢化合物。
在反应过程中,铜基催化剂的作用是吸附二氧化碳分子并将其活化,使其更容易被还原。
同时,铜基催化剂还可以促进氢离子的还原,从而生成有价值的碳氢化合物。
铜基电催化二氧化碳还原的优点在于其高效、环保和可再生性。
与传统的二氧化碳排放控制技术相比,铜基电催化二氧化碳还原不仅可以减少二氧化碳的排放,还可以将排放的二氧化碳转化为有价值的碳氢化合物,从而实现碳资源的循环利用。
此外,铜基电催化二氧化碳还原技术还可以利用可再生能源产生的电能,如太阳能、风能等,进一步降低碳排放。
然而,铜基电催化二氧化碳还原也存在一些挑战和难点。
首先,催化剂的活性需要进一步提高,以降低反应所需的能量和温度。
其次,反应过程中可能会产生多种产物,需要优化催化剂的组成和反应条件,以提高产物选择性和纯度。
此外,反应过程中的副反应和腐蚀问题也需要得到有效控制。
综上所述,铜基电催化二氧化碳还原是一种具有广阔应用前景的技术。
虽然目前该技术还存在一些挑战和难点,但随着科研的不断深入和技术的不断进步,相信铜基电催化二氧化碳还原将会在未来得到更广泛的应用和推广。
1,3-Dipolar cycloaddition of azides with electron-deficientalkynes under mild condition in waterZengmin Li,a,b Tae Seok Seo a,b and Jingyue Ju a,b,*aColumbia Genome Center,Columbia University College of Physicians and Surgeons,New York,NY 10032,USAbDepartment of Chemical Engineering,Columbia University,New York,NY 10027,USAReceived 29September 2003;revised 16February 2004;accepted 17February 2004Abstract—We report a simple synthetic protocol for the 1,3-dipolar cycloaddition of azides with electron-deficient alkynes.Alkyne with at least one neighboring electron-withdrawing group proceeds with the cycloaddition successfully without any catalysts at room temperature in water.Under this simple condition,we evaluated a series of small molecule model reactions and then coupled an azido-DNA molecule with electron-deficient alkynes for the formation of [1,2,3]-triazole heterocycle,providing a potential method for introducing functional groups to DNA under biological conditions.Ó2004Elsevier Ltd.All rights reserved.Since the 1,3-dipolar cycloaddition of azides with alky-nes was investigated by Huisgen et al.1it has attracted much attention because of theoretical interest of the reaction 2and the synthetic importance of the aromatic and nonaromatic five-membered [1,2,3]-triazole hetero-cycles.[1,2,3]-Triazole derivatives have been reported to exhibit antimicrobial activity,3as inhibitors of human leukocyte elastase,4as synthons for the preparation of antitumor dehydropyrrolizidine alkaloids,5and for the modification of nucleosides as antiviral agents.6The traditional method for producing the triazole by cycloaddition requires elevated temperature,typically in refluxing conditions.It is known that alkynes with an electron-withdrawing functional group favor this irre-versible Huisgen cycloaddition of azides and alkynes (Scheme 1).7We previously explored this reaction for site-specific fluorescent labeling of oligonucleotide forDNA sequencing.8Recently,new synthetic methods based on catalysts have been reported for the formation of [1,2,3]-triazoles.Cucurbituril and acetylcholinesterase were employed to lower the activation barrier for the azide–alkyne cycloaddition by sequestering the two components inside a host structure.9;10The copper(I)-catalyzed reaction unites azides and terminal alkynes regiospecifically to give only one-specific regioisomer,the 1,4-disubstituted [1,2,3]-triazole.11This copper(I)-catalyzed system was subsequently applied for the attachment of synthetic oligosaccharides to microtiter plate for biological assayes.12The same reaction was also recently explored for protein and cell surface labeling.13So far,the 1,3-dipolar cycloaddition between the alkynes and azides was conducted either at high temperature thermodynamically or at room temperature catalytically.In the process of optimizing the 1,3-dipolar cycloaddition for DNA analysis and immobilization,we have found that if an electron-deficient internal or terminal alkyne is used,the 1,3-dipolar cycloaddition reaction can be car-ried out successfully using a simple protocol without any catalysts at room temperature in water,which is fully compatible with DNA modification inside a cell under biological conditions.However,under the same condi-tion,alkynes without a neighboring electron-withdraw-ing group do not produce any cycloaddition products.In the first series of experiments,we evaluated the reaction between ethyl 5-azidovalerate and electron-deficient alkynes (Table 1).Keywords :1,3-Dipolar cycloaddition;Electron-deficient alkynes;50-Azido DNA.*Corresponding author.Tel.:+1-212-851-5172;fax:+1-212-851-5176;e-mail:dj222@Scheme 1.0040-4039/$-see front matter Ó2004Elsevier Ltd.All rights reserved.doi:10.1016/j.tetlet.2004.02.089Tetrahedron Letters 45(2004)3143–3146TetrahedronLettersEthyl 5-azidovalerate was readily prepared from ethyl 5-bromovalerate by a nucleophilic substitution with NaN 3in DMSO.14The pure products of cycloaddition were obtained simply by stirring the two reagents in water at room temperature for 6–12h and then isolated by extraction followed by chromatography,or by directly filtering the precipitate followed by recrystallization from ethanol.15We determined the regiochemistry of the entry 3product by X-ray crystallography (Fig.1),indicating that only 1,4-regioisomer was produced selectively,which implies that the cycloaddition inter-mediate was mainly controlled by the steric hindrance effect.To compare the result of the entry 3product with that of the catalyst-mediated reaction,the same product was produced in the presence of CuCl (entry 4).When Cu(I)catalyst was used for cycloaddition with terminal alkynes,the reaction was completed in 1h with a yield of90%,producing a single 1,4-regioisomer identical to the product in entry 3.Similarly,the entry 6product (85%)was produced with a higher yield in the presence of Cu(I)catalyst than that of the entry 5product (67%).However,the Cu(I)catalyzed reaction only works for the terminal alkynes.11Encouraged by these results,we applied this synthetic protocol to the cycloaddition of an azido-DNA with electron-deficient alkynes (Table 2).The azido-labeled DNA was prepared by reacting succinimidyl 5-azido-valerate with an amino-linker modified oligonucleotide (50-amino-GTT TTC CCA GTC ACG ACG-30;M13À40universal forward sequencing primer,m =z 5631)with a 96%yield.The azido-labeled DNA was charac-terized by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI–TOF MS),which showed a single major peak at m =z 5757Da (calcd value:m =z 5756Da).8In a typical reaction,the azido-DNA (4nmol)was reacted with 300-fold excess of alkynyl compounds in 60l L water at room temperature for 48h.After the reaction,excess alkynes were removed by size-exclusion chromatography and the resulting product was desalted with an oligonucleotide purifica-tion cartridge.The product was purified by HPLC and analyzed with MALDI–TOF MS.16The major peaks in the mass spectra generated by entries 7and 8products matched exactly with the calculated values as shown in Figure 2.The product of entry 9was synthesized using the Cu(I)catalyst in 24h with a 60%yield.Figure 1.X-ray structure of the entry 3product (1-ethoxycarbonyl butyl-4-methoxycarbonyl-1,2,3-triazole):orthorhombic;a Âb Âc ,5:6001 A Â6:9381 A Â33:880 A;a ;b ;c ¼90°;90°;90°;R ¼0:0762and R w ¼0:1539.Table 1.Reaction of ethyl 5-azidovalerate with electron-deficient alkynes Entry aAlkyneProductYield (%)1812943824b 905676b 85a For all entries,the azido compound is ethyl5-azidovalerate,rt,water solvent,t R :6–12h.bCuCl (0.1equiv),t R :1h.3144Z.Li et al./Tetrahedron Letters 45(2004)3143–3146In summary,we reported here a simple and mild pro-tocol for the1,3-dipolar cycloaddition between azides and alkynes.When the alkyne(either terminal or internal)has at least one neighboring electron-with-drawing functional group,the triazole formation can be achieved at room temperature in water without any catalysts.In the case of the terminal alkynes,the cycloaddition was proceeded much faster in the presence of Cu(I)catalyst.AcknowledgementsThis work is supported by the National Science Foun-dation(Sensing and Imaging Initiative Grant0097793),and a Center of Excellence in Genomic Science Grant (P50HG002806)from the National Institutes of Health.References and notes1.(a)Huisgen,R.;Szeimies,G.;Moebius,L.Chem.Ber.1967,100,2494–2507;(b)Huisgen,R.1,3-Dipolar Cyclo-addition Chemistry;New York:Wiley,1984;(c)Huisgen, R.Pure Appl.Chem.1989,61,613–628.2.Lwowski,W.In1,3-Dipolar Cycloaddition Chemistry;Wiley:New York,1984;Vol.1.3.Hartzel,L.W.;Benson,F.R.J.Am.Chem.Soc.1954,76,667–670.4.Hlasta,D.J.;Ackerman,.Chem.1994,59,6184–6189.5.Pearson,W.H.;Bergmeier,S.C.;Chytra,J.A.Synthesis1990,156–159.6.Noriis,P.;Horton,D.;Levine,B.R.Heterocycles1996,43,2643–2656.7.(a)Kolb,H.C.;Finn,M.G.;Sharpless,K.B.AngewChem.,Int.Ed.2001,40,2005–2021;(b)Palacios, F.;Retana,A.M.;Pagalday,J.Heterocycles1994,38,95–102.8.Seo,T.S.;Li,Z.;Ruparel,H.;Ju,.Chem.2003,68,609–612.9.(a)Mock,W.L.;Irra,T.A.;Wepsiec,J.P.;Manimaran,.Chem.1983,48,3619–3620;(b)Mock,W.L.;Irra,T.A.;Wepsiec,J.P.;Adhya,.Chem.1989, 54,5302–5308;(c)Krasia,T.C.;Steinke,J.H.G.Chem.Commun.2002,22–23;(d)Tuncel,D.;Steinke,J.H.G.mun.2002,496–497;(e)Tuncel,D.;Steinke,J.mun.2001,253–254.10.Lewis,W.G.;Green,L.G.;Grynszpan,F.;Radic,Z.;Carlier,P.R.;Taylor,P.;Finn,M.G.;Sharpless,K.B.Angew.Chem.,Int.Ed.2002,41,1053–1057.11.(a)Rostovtsev,V.V.;Green,J.G.;Fokin,V.V.;Sharpless,K.B.Angew.Chem.,Int.Ed.2002,41,2596–2599;(b)Tornøe, C.W.;Christensen, C.;Meldal,M..Chem.2002,67,3057–3064.12.Fazio,F.;Bryan,M.C.;Blixt,O.;Paulson,J.C.;Wong,C.J.Am.Chem.Soc.2002,124,14397–14402.13.(a)Speers,A.E.;Adam,G.C.;Cravatt,B.F.J.Am.Chem.Soc.2003,125,4686–4687;(b)Link,A.J.;Tirrell,D.A.J.Am.Chem.Soc.2003,125,11164–11165.Table2.Reaction of an azido-DNA with electron-deficient alkynesEntry a Alkyne Product Yield(%)7458679b60a For all entries,the azido compound is50-azido-DNA,rt,48h,watersolvent.b Methyl propiolate(300equiv),CuI(300equiv),N,N-diisopropylethylamine(300equiv),rt,24h,H2O/CH3CN(4/1volumeratio).Figure2.(A)MALDI–TOF MS spectra of the entry7product(m=z:found,5926;calcd,5926)and(B)the entry8product(m=z:found,5840;calcd,5840).Z.Li et al./Tetrahedron Letters45(2004)3143–3146314514.Khoukhi,N.;Vaultier,M.;Carrie,R.Tetrahedron1987,43,1811–1822.15.General procedure:In a pear shapedflask(25mL)equipped with a magnetic stirring bar,1.2mmol of the alkynyl compound was suspended in H2O(5mL),and then mixed with ethyl5-azidovalerate(0.19g,1.2mmol) vigorously at room temperature for6–12h.After the reaction,the organic layer(entries1,2,and5)was separated by extraction with CH2Cl2.The organic phase was washed with H2O,dried over Na2SO4,and concen-trated.The residue was purified by silica gelflash column chromatography using chloroform as an elute to obtain the pure oily products.In case of entries3and4,the product was precipitated as a white solid in water.The product wasfiltered and recrystallized from EtOH.In case of entries4and6,the same procedure was followed as above except that0.1equiv of CuCl was added in the reaction mixture and the reaction time was1h.The product of entry6was purified by silica gelflash column chromatography to obtain an oily product.The isolated products were characterized by1H and13C NMR and HRMS.Spectral data of the entry1product:1H NMR (400MHz,CDCl3)d4.64(t,2H,J¼9:5Hz),4.52–4.41 (m,4H), 4.19–4.11(q,2H,J¼9:5Hz), 2.38(t,2H, J¼9:7Hz),2.08–1.95(m,2H),1.76–1.65(m,2H),1.44(t, 6H,J¼9:5Hz),1.28(t,3H,J¼9:5Hz);13C NMR(75 MHz,CDCl3)d172.7,160.1,158.4,140.2,129.7,62.8,61.7,60.3,49.9,33.2,29.4,21.5,14.1,13.8.HRMS(FABþ)calcd for C15H24O6N3,342.1665(MþHþ);found, 342.1654.Spectral data of the entry2product:1H NMR (400MHz,CDCl3)d4.20(q,2H,J¼7:1Hz),4.12(q,2H, J¼7:1Hz), 3.28(t,2H,J¼6:5Hz), 2.32(t,2H, J¼7:0Hz),1.97(s,3H),1.72–1.60(m,4H),1.28(t,3H, J¼7:1Hz),1.24(t,3H,J¼7:1Hz);13C NMR(75MHz, CDCl3)d172.8,160.2,158.5,140.3,129.8,61.8,50.0,33.3,33.2,29.5,21.8,14.0.HRMS(FABþ)calcd forC13H22O4N3,284.1610[MþHþ];found,284.1610.Spectral data of the entries3and4product:1H NMR(400MHz,CDCl3)d8.18(s,1H),4.53(t,2H,J¼9:4Hz),4.21(q, 2H,J¼9:5Hz),4.04(s,3H),2.45(t,2H,J¼9:5Hz),2.14–2.04(m,2H), 1.80–1.70(m,2H), 1.35(t,3H,J¼9:5Hz);13C NMR(75MHz,CDCl3)d172.7,161.1, 139.9,127.3,60.5,52.2,50.2,33.2,29.4,21.6,14.1;HRMS (FABþ)calcd for C11H18O4N3,256.1297[MþHþ];found, 256.1310.Spectral data of the entries5and6product:1H NMR(400MHz,CDCl3)d7.96–7.93(m,2H),7.68–7.65 (m,1H),7.58–7.54(m,1H),4.74(d,2H,J¼2:4Hz),4.17–4.10(q,2H,J¼7:2Hz),3.30(t,2H,J¼6:5Hz),2.48(t,1H,J¼2:4Hz),2.34(t,2H,J¼6:5Hz),1.73–1.64(m, 4H,J¼6:4Hz), 1.26(t,3H,J¼6:4Hz);13C NMR (75MHz,CDCl3)d173.5,136.4,134.5,129.7,128.5,75.6,60.7,58.0,51.4,34.0,28.6,22.5,14.5;HRMS(FABþ)calcd for C16H22O5N3S,368.1300[MþHþ];found, 368.1280.The crystallographic data of the entry3product has been deposited at the Cambridge Crystallographic Data Center with the deposition number CCDC220803.16.HPLC analysis:HPLC analysis was carried out on aWaters system consisting of a Rheodyne7725I injector,a 600Controller,Xterra MS C18(4:6Â50-mm)column, and996photodiode array detector.Elution was per-formed by using a linear gradient(8–28%)of methanol ina buffer that consists of8.6mM aqueous triethylamineand100mM hexafluoroisopropyl alcohol(pH8.1)at aflow rate of0.5mL/min with the temperature set at 50°C.Under this condition,the elution time of the entry7product was49.6min and that of the entry8 product was28.2min.MALDI–TOF MS analysis: Mass measurement of oligonucleotides was performed using a Voyager e DE MALDI–TOF mass spectrometer.DNA product(20pmol)was suspended in2l L of 3-hydroxypicolinic acid matrix solution.This mixture(0.5l L)was spotted on a stainless steel sample plate,air-dried,and analyzed.The measurement was taken usinga positive ion mode with25kV accelerating voltage,94%grid voltage and a350ns delay time with internal calibration.3146Z.Li et al./Tetrahedron Letters45(2004)3143–3146。