一致性评价重磅参考资料:(USP1092)溶出度试验的开发和验证(续
- 格式:pdf
- 大小:521.21 KB
- 文档页数:12

⼀致性评价药学申报资料附件2化药仿制药⼝服固体制剂⼀致性评价申报资料要求(征求意见稿)第⼀部分:研究资料信息汇总表(研究综述部分)⼀、⽬录1.1 品种概述1.1.1历史沿⾰(介绍产品的历史沿⾰,简述原研产品情况)1. 1.2批准及上市情况1. 1.3临床信息及不良反应1. 1.4最终确定的处⽅、⼯艺及标准情况1. 1.5⽣物药剂学分类1.2 剂型与产品组成(CDE格式为2.3.P.1)1.3 产品再评价研究(参照CDE资料“3.2.P.2 产品开发”)1.3.1处⽅组成1.3.1.1原料药1.3.1.2辅料1.3.2 制剂的再研发(相对处⽅、⼯艺有改变的品种)1.3.2.1处⽅再研发(如有处⽅改变,详述具体内容)1.3.2.2⽣产⼯艺再研发(如有⼯艺改变,详述具体内容)1.4 ⽣产(参照CDE资料3.2.P.3,删去与注射剂相关的叙述)1.4.1⽣产商1.4.2批处⽅1.4.3⽣产⼯艺和⼯艺控制1.4.4关键⼯艺步骤和中间体的控制1.4.5⼯艺验证和评价1.4.6 临床试验/BE试验样品的⽣产情况1.5 原辅料的控制1.6 包装材料(基本同3.2.P.2.4包装材料/容器,考虑容器主要指注射剂,暂不在题⽬中强调)1.6.1 包装材料类型1.6.2 选择依据1.7 质量控制(基本同CDE资料3.2.P.6制剂的质量控制)1.7.1 质量标准1.7.2 分析⽅法1.7.3 分析⽅法的验证1.7.4 批检验报告1.7.5杂质谱分析1.7.6质量标准制定依据1.8 对照品1.9 稳定性(参照CDE资料“3.2.P.7稳定性”)1.9.1稳定性总结1.9.2后续稳定性承诺和稳定性⽅案(针对有处⽅和⽣产⼯艺改变的品种)1.9.3稳定性数据1.10 参⽐制剂1.10.1参⽐制剂的选择1.10.2基本信息1.10.3质量考察1.10.4溶出曲线考察1.10.5溶出曲线长期稳定性考察1.11 体外评价1.11.1质量⼀致性评价1.11.1.1国内外质量标准收载情况⽐较(含国内外药典标准、橙⽪书等)1.11.1.2关键质量属性研究(影响⼀致性评价的关键参数,例如杂质分析、晶型等)1.11.1.3参⽐制剂与被评价制剂的检验结果1.11.2溶出曲线相似性评价1.11.2.1体外溶出试验⽅法建⽴(含⽅法学验证)1.11.2.2不同溶出仪之间结果差异考察1.11.2.3批内与批间差异考察1.11.2.4溶出曲线相似性⽐较结果1.12 体内评价1.12.1⽣物等效性(按CDE相关资料要求提供)1.12.1.1 ⽤于⽣物等效性试验的样品处⽅及⽣产规模1.12.1.2 不同规格产品的⽣物等效性试验情况1.12.1.3 ⽣物等效性试验设计与实施1.12.1.4 试验受试者1.12.1.5 ⽅案偏离1.12.1.6 安全性评估1.12.1.7 试验结果1.12.1.8 ⽣物样本分析测定1.12.1.9 分析⽅法验证1.12.1.10 质量保证1.12.2 临床有效性1.13 综合评价1.14 参考⽂献1.15附件⼆、信息汇总表正⽂及撰写要求1.1 品种概述1.1.1 历史沿⾰说明同品种原研产品上市背景信息,包括品种治疗领域、国内外上市情况、⽴题的合理性分析。

【重磅推送】USP<1090>体内生物等效性试验指南第二部分本文翻译自USP39-NF34 <1090>Assessment of drug product performance-Bioavailability, Bioequivalence, and Dissolution.溶出度和体外产品性能作为法定物质,USP专论提供了公开的质量标准,包括一系列检查方法,分析用对照以及限度标准。
大多数口服固体制剂,包括口服悬浊液,需要进行溶出度或者药物释放度检查。
药物溶出度和药物释放度检查分别在USP 通则溶出度<711>与释放度<724>章节中有描述。
这些公开的质量标准用来进行质量控制检查以及上市获准。
只有获得管理机构允许时,USP专论中的溶出度检查才与BA及BE相关联。
如果没有这个关联,其将仅仅作为批放行的质量控制检查的方法。
FDA的指导原则包括1.《行业指导原则-速释口服固体制剂溶出度检查Guidance for Industry—DissolutionTesting of Immediate Release Solid Oral Dosage Forms(1977)》(/; 请以文件名检索),2.《行业指导原则-延迟释放制口服制剂:开发、评估及体内外相关性的应用Guidance forIndustry—Extended Release Oral Dosage Forms: Development, Evaluation, andApplication of In Vitro/In Vivo Correlation(1977)》(/; 请以文件名检索)。
溶出度和体外生物利用度药物溶出度和释放度检查在药物制剂开发过程中非常有用,可鉴别关键生产属性如辅料性质、生产工艺等对药物制剂特性的影响。
在药物开发过程中,需要确定最优溶出度条件以辨别药物制剂处方及生产工艺变更。

药学一致性评价中的体外溶出和质量研究刘振, 分析服务部高级总监September, 20162概要体外溶出和质量研究的一般考虑 经验分享和实例分析药明康德一体化的分析服务平台药学一致性评价分析研究法规要求和流程1普通口服固体制剂溶出度试验技术指导原则•20150205总局关于发布普通口服固体制剂参比制剂选择和确定等3个技术指导原则的通告(2016年第61号)•20160318总局关于发布药物溶出度仪机械验证指导原则的通告(2016年第78号)•20160429总局关于发布人体生物等效性试验豁免指导原则的通告(2016年第87号)•20160519总局关于发布化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)的通告(2016年第120号)•2016081634No.标题CTD 编号 8.4 原辅料的控制 3.2.P.4 8.5 制剂的质量控制3.2.P.5 8.5.1 质量标准 3.2.P.5.1 8.5.2 分析方法3.2.P.5.2 8.5.3 分析方法的验证 3.2.P.5.3 8.5.4 批检验报告 3.2.P.5.4 8.5.5 杂质谱分析3.2.P.5.5 8.5.6 质量标准制定依据 3.2.P.5.6 8.6 对照品 3.2.P.6 8.7 包装材料 3.2.P.7 8.8 稳定性3.2.P.8 8.8.1 稳定性总结3.2.P.8.1 8.8.2 后续稳定性承诺和稳定性方案 3.2.P.8.2 8.8.3 稳定性数据 3.2.P.8.39 参比制剂 9.3 质量考察 9.4 溶出曲线考察9.5 溶出曲线稳定性考察 10 质量一致性评价 10.1 质量标准比较10.2 关键质量属性研究10.3 参比制剂与被评价制剂的检验结果 11 溶出曲线相似性评价11.1 体外溶出试验方法建立(含方法学验证) 11.2 批内与批间差异考察 11.3溶出曲线相似性比较结果分析数据汇总调研: 各国药典、标准、数据库 分析方法开发验证: 主要包括溶出曲线、有关物质体外溶出曲线对比检测 批内批间差异考察、相似性评价 质量标准研究和对比检测 杂质谱分析,关键质量属性 稳定性试验 参比制剂和试验制剂5概要经验分享和实例分析药明康德一体化的分析服务平台药学一致性评价分析研究法规要求和流程 体外溶出和质量研究的一般考虑26溶出分析方法开发和溶出曲线对比方法需求:选择性/灵敏度/准确度/精密度(VOC )方法开发:满足阶段性的需求和使用目的)风险评估 :耐用性,重现性 方法确定:区分定义关键方法性能影响参数方法验证 持续优化和变更指导药物制剂的研发,评价仿制制剂的质量,溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临床疗效差异的风险。


(1092)溶出度试验的开发和验证【中英文对照版】INTRODUCTION前言Purpose目的The Dissolution Procedure: Developmentand Validation <1092> provides a comprehensive approach covering items to considerfor developing and validating dissolution procedures and the accompanyinganalytical procedures. It addresses the use of automation throughout the testand provides guidance and criteria for validation. It also addresses thetreatment of the data generated and the interpretation of acceptance criteriafor immediate- and modified-release solid oral dosage forms.溶出实验:开发和验证(1092)指导原则提供了在溶出度方法开发和验证过程中以及采用相应分析方法时需要考虑的因素。
本指导原则贯穿溶出度实验的全部过程,并对方法提供了指导和验证标准。
同时它还涉及对普通制剂和缓释制剂所生成的数据和接受标准进行说明。
Scope范围Chapter <1092> addresses the development andvalidation of dissolution procedures, with a focus on solid oral dosage forms.Many of the concepts presented, however, may be applicable to other dosageforms and routes of administration. General recommendations are given with theunderstanding that modifications of the apparatus and procedures as given in USP general chapters need to be justified.<1092>章节讨论了溶出度实验的开发和验证,重点是口服固体制剂。
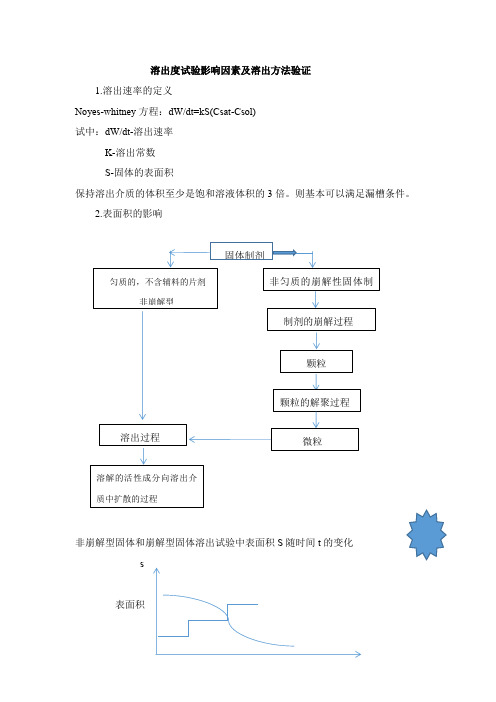
溶出度试验影响因素及溶出方法验证1.溶出速率的定义Noyes-whitney 方程:dW/dt=kS(Csat-Csol) 试中:dW/dt-溶出速率 K-溶出常数 S-固体的表面积保持溶出介质的体积至少是饱和溶液体积的3倍。
则基本可以满足漏槽条件。
2.表面积的影响非崩解型固体和崩解型固体溶出试验中表面积S 随时间t 的变化 s表面积溶出度仪的影响因素1.晃动的影响A.与TIR值小于2.0mm的结果相比,晃动偏差TIR值为1.0~2.0mm,水杨酸片和泼尼松片的溶出量分别增加约5%。
B.溶出度仪设计中需注意两个因素1.要求转轴必须垂直;2.转轴应有两个固定点,仪器顶部到转轴卡盘的距离至少应不低于卡盘至转篮或桨叶的距离。
2.转轴的直线度a.转轴的直线度是控制晃动的关键仪器指标,应确保其直线度b.溶出度仪的涉及要求桨叶或篮体的顶端距卡盘的距离至少要6英寸(15.2cm)。
3.其它搅拌装置的变动因素A.篮杆和桨杆是精密部件,使用时应小心,这些精密仪器在实验室的抽屉中存放时,会破坏不锈钢桨表面的。
引起弯曲和变性。
应有适当的支架供转轴存放。
B.桨叶应没有锐角。
尖锐部分会引起涡流而不是层流。
取用或放置篮时只能接触篮的上部边框,随着时间的增加,特别是在酸性介质时,筛网的孔径会有变化。
可用放大镜检,必要时需更换篮。
4.振动4.1振动的来源A实验室中能产生振动的仪器,包括通风橱和离心机,空调,风扇,离心机等。
B.人员走动,关门,开门。
C.早期水浴加热和溶出仪连在一起,目前的溶出度仪设计都采用外置循环泵与水浴连接。
4.2应将盛有溶出介质的溶出杯的各种来源的振动水平降低到0.1mil。
5.搅拌装置的准直度转轴的轴线与溶出杯的中心轴线间的偏离和倾斜对溶出介质流体动力学影响严重,可使溶出结果差异达到±25%。
影响结果很明显。
6.溶出杯中转轴的中心度7.搅拌速度规定转速一般不得过4%,转速的变化对溶出速率的影响几乎是线性的。
麻烦帮忙翻译USP35中一下内容:(编号为页码)<11>USP对照标准品<21>温度计<31>容量仪器<41>重量与天平<429>粒度的光衍射测定<467>残留溶剂<616>松装密度/堆密度和振实密度<621>色谱分析法<641>溶液的澄清度<699>固体密度<724>药物释放度(P442)-2<786>通过分析筛选估计粒度分布/通过筛分法估算粒子分布<791>pH<811>粉体细度/粉剂细度<851>分光光度法与光散射(P577)-6<905>含量均匀度(P593)-3<921>水分测定<1051>清洗玻璃容器(P754)<1087>内部的溶出度(P871)-3<1092>溶出程序:开发与验证(P889)<1216>片剂的脆碎度(P1120)<1217>片剂破碎力<1251>在分析天平上称量(标红的部分为已翻译完成)<197>分光光度鉴别测试(P196)红外吸收干燥待测样品和分析用对照品的制备有7种方法。
各论中引用<197K>意味着待测物质与溴化钾完全混合。
各论中引用<197M>意味着待测物质经过精细的磨碎,并溶于矿物油中。
各论中引用<197F>意味着待测物质悬浮在不同的盘中(plate)(如,氯化钠或溴化钾)。
各论中引用<197S>意味着制备指定浓度的溶液,且溶解于各个各论中指定的溶剂中,除非各个各论中对光程长吸收池(cell path length)另有规定,否则溶液的检测应在0.1mm吸收池(cell)中进行。
各论中引用<197A>意味着待测物质与内反射元件亲密接触,用于衰减全反射(ATR)分析。
圣诞礼物 一致性评价重磅参考资料:(USP1092)溶出度试验的开发和验证(续)20151225刘建华医药信息新药开发3.分析整理溶出步骤也是一个复杂的样品制备过程。
用于测定溶出过程中药物溶出量的处理和分析过程,称为“分析整理”。
虽然本章讨论的分光光度法和高效液相色谱法是最常用的分析方法,其他适宜的分析技术也可以使用。
在第5节,将详细描述方法验证标准。
3.1 样品处理溶出样品在取样后,需要进一步的处理,使能够满足样品释放量的分析方法的测定要求。
例如,过滤可用于除去未溶解的颗粒物样品,或者避光、冷藏贮存样品。
此外,样品可能需要稀释至方法线性范围内的测定浓度。
使用高效液相色谱法时,尽可能采用流动相稀释至样品以减少溶出介质对样品测定的影响。
根据产品特性的要求,其他类型的处理方式也是存在的。
例如加入适当的试剂使产生干扰的物质消除或者失活。
然而,分离可能是不可能的或需要的不是必需的,在一些情况下,在原位测量的方法,如纤维光学或电化学测定方法可能是有用的。
3.2 过滤在上面1.1章节中已经讲述。
3.3 离心离心处理样品是不优选的,具体原因有以下几个方面:在固体颗粒除去之前,药物溶解可以继续发生,是药物的溶出浓度增大,并且离心的动力也可能导致增加溶解的药物颗粒。
然而当所有常见滤膜对药物均有吸附或者所有滤膜均干扰药物的测定时(例如,使用荧光定量),可以选择离心法处理样品。
离心法可以证明是有用的,在方法开发的过滤材料的适用性评价。
3.4 分析方法用于溶出度测定的常用分析方法一般为分光光度法或液相色谱法。
分光光度法较高效液相法更简便快捷,并且分光光度法较HPLC法更容易自动化,并且溶剂量使用较少。
但是分光光度法测定需要专属性良好。
高效液相色谱法是首选的原因有很多,如提供较宽测定范围,减少了需要稀释样品的步骤,提高了低浓度样品的分析灵敏度,并且可用于辅料或者多组分互相干扰的样品的测定。
目前的高效液相色谱系统采用自动进样器,提高了自动化。
苯磺酸氨氯地平片体外溶出度一致性评价及体内生物等效性研究池王胄胡旭华(上海天慈国际药业有限公司上海 201315)摘要目的:考察苯磺酸氨氯地平片体外溶出曲线相似性与体内生物等效性,以评估国产制剂与参比制剂的质量一致性。
方法:建立体外溶出测定方法以评价在不同溶出介质中国产制剂与参比制剂的溶出一致性;同时,在60名中国健康成年志愿者中进行临床体内生物等效性评价。
结果:在体外不同溶出介质中待评价的两种片剂累计溶出均大于85%;生物等效性中餐前餐后的药峰浓度、药时曲线下面积的几何均值比的90% CI均在92%~104%内。
结论:苯磺酸氨氯地平片国产制剂产品质量与参比制剂具有一致性,且粉末直压法工艺简单,降低了生产成本,提高了生产效率。
关键词苯磺酸氨氯地平 体外溶出度 生物等效试验中图分类号:R927.11; R972.4 文献标志码:A 文章编号:1006-1533(2023)23-0107-04引用本文池王胄, 胡旭华. 苯磺酸氨氯地平片体外溶出度一致性评价及体内生物等效性研究[J]. 上海医药, 2023, 44(23): 107-110.In vitro dissolution consistency evaluation and in vivo bioequivalence studieson amlodipine besylate tabletsCHI Wangzhou, HU Xuhua(Shanghai Tianci International Pharmaceuticals Co., Ltd., Shanghai 201315, China)ABSTRACT Objective: To investigate the similarity of the in vitro dissolution curves and in vivo bioequivalence ofamlodipine besylate tablets and to evaluate the quality consistency between homemade amlodipine besylate tablet and its reference listed drug (RLD). Methods: An in vitro dissolution determination method was established to evaluate the consistency of the dissolution between homemade tablets and RLD in different dissolution media. At the same time, in vivo bioequivalence evaluation was carried out in 60 healthy adult volunteers in China. Results: The cumulative in vitro dissolution of the two tablets to be evaluated in different dissolution media was greater than 85%. The bioequivalence results showed that the 90% CI for geometric mean ratios of C max and AUC0-72 h on an empty stomach and after meals was within 92% to 104%. Conclusion: The product quality of amlodipine besylate tablets is consistent with that of RLD, and the powder direct compression method is simple, which can reduce production cost and improve production efficiency.KEY WORDS amlodipine besylate; in vitro dissolution; bioequivalence苯磺酸氨氯地平片属于外周动脉血管扩张剂,适用于高血压患者(单独用药或与其他抗高血压药物合用)和慢性稳定性心绞痛及变异型心绞痛患者(单独用药或与其他抗心绞痛药物合用),机理是作用在血管平滑肌来降低外周血管阻力和血压[1-2]。
(完整版)USP-1092-溶出度试验的开发和验证(中英文对照版).docx( 1092)溶出度试验的开发和验证【中英文对照版】INTRODUCTION前言Purpose目的The Dissolution Procedure: Developmentand Validation<1092>provides a comprehensive approach covering items to considerfor developing and validating dissolution procedures and the accompanyinganalytical procedures. It addresses the use of automation throughout the testandprovides guidance and criteria for validation.It also addresses thetreatment of the data generated and the interpretation of acceptance criteriafor immediate-and modified-releasesolid oral dosage forms.溶出实验:开发和验证(1092)指导原则提供了在溶出度方法开发和验证过程中以及采用相应分析方法时需要考虑的因素。
本指导原则贯穿溶出度实验的全部过程,并对方法提供了指导和验证标准。
同时它还涉及对普通制剂和缓释制剂所生成的数据和接受标准进行说明。
Scope范围Chapter<1092>addresses the development andvalidationof dissolution procedures,with a focus on solid oral dosage forms.Many of the concepts presented, however, may be applicable to other dosageforms and routes of administration. General recommendations are given with theunderstandingthat modifications of the apparatus and procedures as given in USP general chapters need to be justified.<1092>章节讨论了溶出度实验的开发和验证,重点是口服固体制剂。
圣诞礼物 一致性评价重磅参考资料:(USP1092)溶出度试验的开发和验证(续)20151225刘建华医药信息新药开发3.分析整理溶出步骤也是一个复杂的样品制备过程。
用于测定溶出过程中药物溶出量的处理和分析过程,称为“分析整理”。
虽然本章讨论的分光光度法和高效液相色谱法是最常用的分析方法,其他适宜的分析技术也可以使用。
在第5节,将详细描述方法验证标准。
3.1 样品处理溶出样品在取样后,需要进一步的处理,使能够满足样品释放量的分析方法的测定要求。
例如,过滤可用于除去未溶解的颗粒物样品,或者避光、冷藏贮存样品。
此外,样品可能需要稀释至方法线性范围内的测定浓度。
使用高效液相色谱法时,尽可能采用流动相稀释至样品以减少溶出介质对样品测定的影响。
根据产品特性的要求,其他类型的处理方式也是存在的。
例如加入适当的试剂使产生干扰的物质消除或者失活。
然而,分离可能是不可能的或需要的不是必需的,在一些情况下,在原位测量的方法,如纤维光学或电化学测定方法可能是有用的。
3.2 过滤在上面1.1章节中已经讲述。
3.3 离心离心处理样品是不优选的,具体原因有以下几个方面:在固体颗粒除去之前,药物溶解可以继续发生,是药物的溶出浓度增大,并且离心的动力也可能导致增加溶解的药物颗粒。
然而当所有常见滤膜对药物均有吸附或者所有滤膜均干扰药物的测定时(例如,使用荧光定量),可以选择离心法处理样品。
离心法可以证明是有用的,在方法开发的过滤材料的适用性评价。
3.4 分析方法用于溶出度测定的常用分析方法一般为分光光度法或液相色谱法。
分光光度法较高效液相法更简便快捷,并且分光光度法较HPLC法更容易自动化,并且溶剂量使用较少。
但是分光光度法测定需要专属性良好。
高效液相色谱法是首选的原因有很多,如提供较宽测定范围,减少了需要稀释样品的步骤,提高了低浓度样品的分析灵敏度,并且可用于辅料或者多组分互相干扰的样品的测定。
目前的高效液相色谱系统采用自动进样器,提高了自动化。
3.5 光谱分析直接分光光度法分析可以采用手动操作。
另外也可以采用自动吸样系统或者流通进样池进行自动化分析操作。
按照标准操作规程或者计量文件的要求进行仪器日常的仪器检查,清洁和维护,有助于确保仪器的准确运行。
分光光度计的比色皿的长度一般为0.02cm~1cm,如果测定浓度较小的样品也可以使用长度较大的比色皿,较长比色皿中存在气泡可能引起仪器错误。
细胞排列和气泡可能是错误的来源。
较短的比色皿可以使样品不用稀释直接进行测定,然而不管使用什么比色皿,样品溶液的线性范围以及标准误差必须进样验证。
检测波长选择必须基于样物溶液吸收光谱。
在某些情况下,药物在溶出介质中降解(例如,含阿司匹林的制剂),一般会选取其降解物一致的吸收波长。
对于辅料有干扰的情况,可以选用多波长进行分析:吸光度从赋形剂在分析的波长可以通过波长处吸光度最小的药物从吸收度比值确定。
在方法经过验证后,对照品溶液浓度一般只有一个浓度值,无论选取溶出量为100%或者溶出度限度(Q)值均是可以的,因为线性范围已经经过验证。
但是在验证试验之前,无论溶出度分析曲线的测定,或者产品的优势分析,均需要使用多个对照品进行测定,已涵盖了预对照品溶液和样品溶液均应在溶出的线性浓度范围内采用相同波长进行测量。
然而,少量的有机溶剂可用于制备的对照品溶液,以满足在验证过程中测定的准确度。
吸光系数α的计算公式如下(朗伯比尔定律):α=A/bcA=吸光度b=比色皿长度(cm)c=浓度(mg/ml)用于溶出试验吸光度的单位通常为AU•毫升/毫克,其中AU是吸收度单位,.历史的资料可用来提供分析物可接受的吸收范围的(使用适当的路径长度单元)。
这个值可能会在故障排除异常数据时非常有用。
纤维光学作为采样测定方法,通过适当的验证,是一种选择。
3.6 HPLC法对于HPLC分析,由注射溶解介质对色谱图的影响需要列出。
如果目标峰响的应解决不好,溶剂的较大干扰可能影响响应的准确度和精确。
如果需要进样较大量(>100毫升)这更为重要。
系统适用性试验可评估峰形、溶剂干扰、和主峰相邻峰的分离、和进样精密度。
至少,该精密度是至关重要的。
理想情况下,标准溶液应用溶解介质进行稀释,要在该方法的线性范围内的浓度,例如,100%,或所选择的制剂剂量值稀释。
然而,有机溶剂可以在制剂标准中使用,在验证过程中只要满足该精密度标准。
在一些情况下,为了改善峰形,样品可以用流动相稀释,以改善峰形。
在线性浓度范围内制备标准溶液和样品溶液应,并在同一波长处测定。
4.自动化溶出度测定的自动化有很多方式和层级。
溶出准备、开启、设置取样点和取样以及清洗均能实现自动化。
完全自动化是指每个操作步骤均实现自动化,比如溶出介质的配制或者取样。
本章节主要讨论可以实现自动化的步骤。
自动化的水平取决于溶出仪是开环的还是闭环,同时也取决于分析仪器是脱机的还是联机的。
联机分析是指测试样品能够输送到测定系统中并返还到溶出仪中,例如含有流通比色皿的分光光度计。
脱机分析是指将样品去除溶出仪,然后进行后续的分析(最典型的为高效液相色谱法),分析过程中消耗掉样品。
仪器配置的选择取决于样品的量以及分析所限制的时间。
自动化操作需要报告药典规定的仪器仪器偏差,如清洗或者更换介质遗留在管路中的残留,并且非药典标准化操作的步骤必须进行验证。
指导原则(711)规定的标准程序的偏差,如使用采样探头或者光纤探头,必须按照标准程序进行验证。
4.1介质的配制自动化配制溶出介质一般是通过稀释已经配制完的浓溶液完成。
自动配置溶出介质程序一般是通过检测重量或者体积将介质输送到溶出杯中。
浓缩液物理化学稳定性同使用过程中稀溶液的均一性一样是一个重要的问题,并且必须进行考察。
缓冲溶液和含有表面活性剂的溶出介质可能会存在稳定性问题,例如化学降解或者pH值的改变。
物理稳定性主要是沉淀、重结晶或者相分离,也应该防止发生。
如果介质要求脱气,脱气水平应该符合规定。
介质中氧气的含量可以通过上述章节2.1脱气程序的方法进行评估。
4.2 样品的取样时间溶出杯中应有一个循环的取样管路。
自动取样和回收大大方便手动取样,因为它减少了取样过程中所引起的变量。
同时在早期取样点手动取样很难满足药典要求的取样时间相对标准偏差在±2%的要求。
4.3 取样和过滤自动化是代替手动取样的一种实用的方法,尤其是对于多个取样点的试验。
样品的取样和过滤使用弯管接头或者一系列步骤从溶出仪中直接到测定仪器中。
样品溶液从溶出仪中取走后,可以不回收至溶出仪中(消耗取样)或者返回至溶出仪中(循环取样)。
目前有许多家品牌的自动溶出仪,有半自动的也有全自动的。
仪器应当按照相应标准操作规程进行日常的性能检查、清洗和维护,以满足仪器正常运行。
在整个取样过程中取样针可以保持在或者不保持在溶出杯中。
取样针或者光纤取样针可能会影响整体管路的流体学,因此需要进行充足的验证以保证不对药物的溶出速率造成重大的影响。
如果使用的滤膜与手动操作的不同,同样需要对不同的滤膜分别经行考察。
药典设备1和设备2的采样区的位置是中途从搅拌元件顶部到介质表面,根据介质的体积,取样针应从取样区进行取样。
用于通过空心轴进行取样的仪器,应采用一种方法来调整入口孔的深度以使其符合要求。
取样量取决于管路、比色皿和其他设备的死体积,必须进行相应的调整。
循环采样可以通管路在取样后将溶液排入溶出杯中或者在两个取样点中间将溶液排入溶出杯中。
在后者的情况下,死体积和浓度效应的影响是重要的考虑因素。
在多个采样时间点的试验中,补液是需要考虑的。
当取样量的总体积超过溶出介质总体积的1%时,影响是很大的。
如果没有循环利用介质,那么应该计算结果,具体见2.5节数据处理。
当后续样品受残留或先前取样的条件的影响,可能会发生交叉污染;第一个样品或条件的影响传递到第二样品。
在处理液体时,在样品溶液中的先前液体的残留物可能污染后续样品溶液。
溶出介质包含表面活性剂或脂质可能会出现一些问题。
根据清洗方案在多个时间点测试和在测试开始以及连续采样都可能发生残留。
这个问题将在4.4节清洁讨论。
溶解的原料药和取样的相互作用和设备转移是重要考虑的,当溶解的原料药发生吸附时,它最经常涉及的是溶解装置或抽样滤波器和管道的表面上。
溶解的原料药在荷电时的吸附可以是pH依赖性的。
溶解的原料药到采样装置部件的吸附应当使用的已知浓度的典型样品溶液(从制剂或原料和辅料混合组分中溶出的样品)进行评估。
通常使用等分的同一样品溶液通过和绕过取样装置(包括采样探头,过滤器,管道,阀门和泵)设计一个交叉验证试验。
有可能对任何种类的材料或设备结构(例如,玻璃或特定的聚合物)的有限选择不能给出建议。
参见5.7自动化注意事项的详细信息。
除了在2.4.3取样部分的信息外,泵和管道的连接处可能是自动化系统污染的来源。
通常用于溶出试验的分光分析程序的干扰,较少关注的。
但是,被研究的制剂含有低剂量的金属盐,如做一些膳食补充剂必须对干扰进行评估。
液体转移通常是通过聚合物管进行。
惰性材料如聚四氟乙烯(PTFE),因为它们的机械性能的影响有时也无法使用。
其中,软管是必需的,例如在蠕动泵或用于在小半径环绕,聚丙烯(PP)或高密度聚乙烯(HDPE)也可以是优选的材料。
取决于聚合物的类型和它的结晶度和密度、主要增塑剂,可能产生组分的浸出。
溶出物可能与分析程序干扰。
被过滤到样品溶液中的浓度通常取决于表面、温度、暴露时间,流体动力学条件和溶出介质的组成。
4.4清洗除了2.4.4清洁部分的信息,自动系统有具体的清洁问题。
例如,推荐在采样时间和内运行时间评估清洗和冲洗的有效性。
在测试之间评估清洁过程也是很重要的。
4.5操作软件和计算的结果根据21 CFR11(17)仪器操作的软件系统和数据评估必须进行验证。
4.6药典程序的常见偏差需要进行验证从药典程序中的一些常见偏差包括:•主轴开始旋转投入样品引起的偏差•停留时间和采样探头位置•循环与消耗采样•在消费采样样品体积置换。
5.验证本章节所涉及的验证是一些典型的验证,但不是包括所有的验证,这些验证参考药典的验证<1225>或者ICH分析方法的验证。
本章节讨论的溶出试验的验证包括两个部分,即溶出过程的验证和数据分析的验证。
溶出过程是指药物在溶出介质中的溶出和取样,数据分析的定义详见第3章节分析整理。
数据分析的验证包括专属性、线性和范围、精密度、准确定/回收率、耐用性和对照品溶液和供试品溶液的稳定性。
而溶出过程的验证主要是评估供试品溶液制备的精密度和耐用性。
数据分析的验证一般使用对照品溶液或者空白辅料溶液或者按照<1225>准确度试验中适用标准加入法制备的溶液进行验证,详细见下面章节。
而溶出过程的验证需要使用有良好特性的产品(例如:比如具有良好的含量均匀度和溶出均一性)。