PCR示意图
- 格式:doc
- 大小:4.84 MB
- 文档页数:12

设计的目的是在两个目标间取得平衡:扩增特异性和扩增效率。引物分析软件将试图通过使用每一引物设计变化的预定值在这两个目标间取得平衡。设计引用有一些需要注意的基本原理:
①引物长度
一般引物长度为18~30碱基。总的说来,决定引物退火温度(Tm值)最重要的因素就是引物的长度。有以下公式可以用于粗略计算引物的退火温度。
在引物长度小于20bp时:[4(G+C)+2(A+T)]-5℃
在引物长度大于20bp时:62.3℃+0.41℃(%G-C)-500/length-5℃
另外有许多软件也可以对退火温度进行计算,其计算原理会各有不同,因此有时计算出的数值可能会有少量差距。为了优化PCR反应,使用确保退火温度不低于54℃的最短的引物可获得最好的效率和特异性。
总的说来,每增加一个核苷酸引物特异性提高4倍,这样,大多数应用的最短引物长度为18个核苷酸。引物长度的上限并不很重要,主要与反应效率有关。由于熵的原因,引物越长,它退火结合到靶DNA上形成供DNA聚合酶结合的稳定双链模板的速率越小。
② GC含量
一般引物序列中G+C含量一般为40%~60%,一对引物的GC含量和Tm值应该协调。若是引物存在严重的GC倾向或AT倾向则可以在引物5’端加适量的A、T或G、C尾巴。
③退火温度
退火温度需要比解链温度低5℃,如果引物碱基数较少,可以适当提高退火温度,这样可以使PCR的特异性增加;如果碱基数较多,那么可以适当减低退火温度,是DNA双链结合。一对引物的退火温度相差4℃~6℃不会影响PCR的产率,但是理想情况下一对引物的退火温度是一样的,可以在55℃~75℃间变化。
④避免扩增模板的二级结构区域
选择扩增片段时最好避开模板的二级结构区域。用有关计算机软件可以预测估计目的片段的稳定二级结构,有助于选择模板。实验表明,待扩区域自由能(△G)小于58.6lkJ/mol时,扩增往往不能成功。若不能避开这一区域时,用7-deaza-2’-脱氧GTP取代dGTP对扩增的成功是有帮助的。

〔PCR的循环参数〕
1、预变性(Initial denaturation).
模板DNA完全变性对PCR能否成功至关重要,一般95℃加热3-5分钟。
2、引物退火(Primer annealing)
退火温度一般需要凭实验(经验)决定。
退火温度对PCR的特异性有较大影响。
Tm值(解链温度)=4(G+C)+2(A+T)
复性温度=Tm值-(5~10℃)
在Tm值允许范围内, 选择较高的复性温度可大大减少引物和模板间的非特异性结合, 提高PCR反应的特异性。复性时间一般为30~60sec,足以使引物与模板之间完全结合。
3、引物延伸
引物延伸一般在72℃进行(Taq酶最适温度)。
延伸时间随扩增片段长短而定。
PCR反应的延伸温度一般选择在70~75℃之间,常用温度为72℃,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1Kb以内的DNA片段,延伸时间1min是足够 的。3~4kb的靶序列需3~4min;扩增10Kb需延伸至15min。延伸进间过长会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。
4、循环中的变性步骤
循环中一般95℃,30秒足以使各种靶DNA序列完全变性:
变性时间过长损害酶活性,过短靶序列变性不彻底,易造成扩增失败。
每完成一个循环需2~4分钟
5、循环数
大多数PCR含25-35循环,过多易产生非特异扩增。
6、最后延伸
在最后一个循环后,反应在72℃维持5-15分钟.使引物延伸完全,并使单链产物退火成双链。
PCR反应的延伸温度一般选择在70~75℃之间,常用温度为72℃,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1Kb以内的DNA片段,延伸时间1min是足够 的。3~4kb的靶序列需3~4min;扩增10Kb需延伸至15min。延伸进间过长会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。

Tm值就是DNA熔解温度,指把DNA的双螺旋结构降解一半时的温度。不同序列的DNA,Tm值不同。DNA中G-C含量越高,Tm值越高,成正比关系。
影响Tm值的因素
①DNA碱基组成:C-G含量越多,Tm值越高;A-T含量越多,Tm值越低。
②溶液的离子强度:在低离子强度中,Tm较低,而且解链的温度范围较宽;在高离子强度中,Tm值较高,解链的温度范围较窄。
③PH值:溶液的PH值在5~9范围内,Tm值变化不明显,当PH>11或PH<4时,Tm值变化明显。
④变性剂:各种变性剂主要是干扰碱基堆积力和氢键的形成而降低Tm值。
⑤DNA双链本身的长度。
溶解曲线相关问题:
这个一般是用SYBY Green作为荧光染料的时候需要做的工作
因为SYBY Green染料是非特异的染料只要有扩增染料就可以镶嵌在双链
中发出荧光。我们做溶解曲线是为了观察我们扩增发出荧光的片段是不是我们需
要的产物。如果溶解曲线做出来只有单峰且出锋位置是你的退火温度那么这
个是你的产物发出的荧光实验结果有效。如果出现多锋或退火温度不对那么
这个实验结果是不可信的你需要再次调整

pcr技术的知识点
PCR,全称为聚合酶链式反应(Polymerase Chain Reaction),是一种在体外扩增DNA分子的技术。自从其产生以来,PCR技术在生物学、医学等领域都扮演着重要角色。本文将介绍PCR技术的基本原理、主要步骤及其应用。
一、PCR技术的基本原理
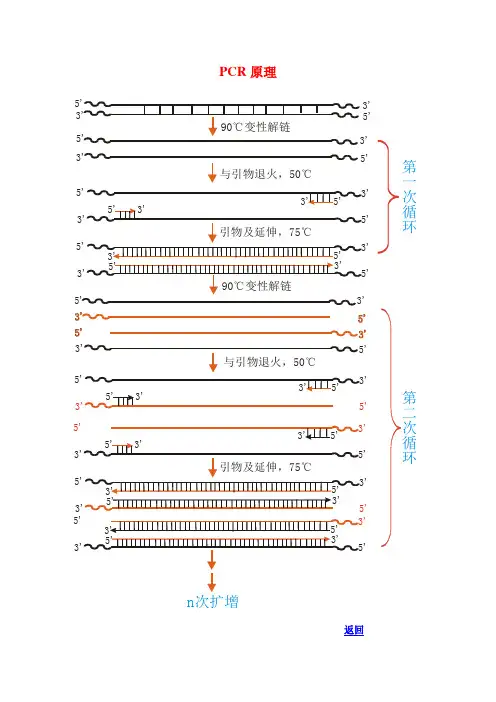
PCR技术是利用DNA聚合酶能够在适宜条件下从单链DNA模板向其补体的方向合成新的互补双链DNA的特性,通过体外操作使目标DNA区段被扩增的一种技术。PCR技术相当于“放大器”,它把少量的DNA分子扩增成数百万分子,这种扩增是平凡的、快速的和特异的。
PCR从样本中扩增一段特定目的序列的DNA,应该是由以下三个部分所组成:一段待扩增的DNA序列,一对荧光探针(引物)和一种特殊的聚合酶。
PCR反应主要可分为以下三个阶段:
1. 解旋(Denaturation):将待扩增的DNA序列在高温条件下变性,形成两条单链DNA模板。
2. 引物与合成(Annealing and Extension):在低温条件下,引物与模板DNA的互补匹配,并由聚合酶在其5'-3'方向上合成新的DNA链。
3. 延伸(Extension):利用DNA聚合酶在适宜温度下合成新的互补双链DNA,形成两个完全相同的分子。
通过不断的循环这三个阶段,从而使得PCR反应体系中的待扩增物质呈指数级别的增长。
二、PCR技术的主要步骤
PCR主要分为以下几个步骤:
1. 样品获取
PCR技术对样品数量和质量要求较高。通常采用的是细胞、肝脏、皮肤、血液等组织或器官中提取DNA分子作为扩增目标。同时,需要注意样品的保存和处理以避免DNA降解。
2. DNA提取
DNA提取技术是PCR技术的基础。不同的样品需要采用不同的DNA提取方法,常见的DNA提取方法有基于酸性、碱性或复合的物理或化学方法等。
3. 引物设计
PCR扩增需要引物特异性和长度适宜,且引物与待扩增的DNA序列互补匹配度高,通常由专业的引物设计软件完成。