各种酶切位点的保护碱基 引物设计必看
- 格式:docx
- 大小:15.72 KB
- 文档页数:6

PCR设计引物时酶切位点的保护碱基引物设计是PCR实验的关键步骤之一,引物的好坏会直接影响到PCR反应的成功与否。
而在引物设计过程中,酶切位点的保护碱基是需要考虑的重要因素之一在PCR实验中,引物的作用是指定PCR反应的放大区域,并提供启动位点供聚合酶结合。
一般情况下,引物至少需要包含一段特定的DNA序列,以便与目标序列互补配对。
在引物设计过程中,选择合适的酶切位点是十分必要的。
酶切位点是指位于特定DNA序列上的限制酶可以识别并切割的区域。
酶切位点的选择通常需要考虑如下几个方面:1.切割效果:选择切割效果好的酶切位点可以提高PCR反应的特异性和灵敏度。
经典的选择是选择一种具有4-6个碱基的酶切位点,并且该位点在引物中间的位置。
这可以有效防止酶切位点的保护碱基对PCR反应的影响。
2.特异性:引物需要选择适合的酶切位点,以确保只有目标序列被放大,而不包括其他与之相关的非特异性序列。
因此,在选择酶切位点时应尽量避免与其他非特异性序列存在相似性。
3.引物长度:引物长度的选择也与酶切位点相关。
如果引物长度过短,可能会导致酶切位点过于靠近PCR反应产物的端点,从而使切割效果不佳。
因此,在引物设计时,应选择适当的引物长度,以保证酶切位点的保护碱基不会对PCR反应产物的生成产生不利影响。
酶切位点的保护碱基是指在特定的DNA序列上,通过选择相应的碱基来避免受到酶切的影响。
常见的保护碱基有甲基化碱基、磷酸化碱基以及接上阻断扩增的非互补碱基等。
1.甲基化碱基:将酶切位点中的一些碱基进行甲基化处理,可以有效地阻止特定酶的切割作用。
甲基化碱基可以通过DNA甲基转移酶进行甲基化修饰。
2.磷酸化碱基:磷酸化碱基是在引物设计过程中添加磷酸基团的方法,通过给酶切位点添加一个磷酸基团来阻断酶的切割作用。
3.非互补碱基:为了阻断酶切位点的切割作用,可以在酶切位点的周围引入一个与其不互补的碱基序列。
这样可以阻断酶的结合和切割。
总的来说,选择合适的酶切位点和保护碱基对PCR实验的成功至关重要。
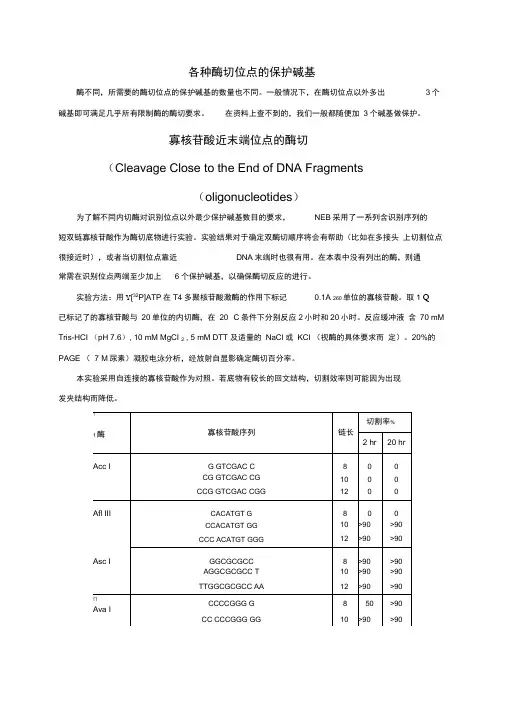
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用Y[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A 260单位的寡核苷酸。
取1 Q已标记了的寡核苷酸与20单位的内切酶,在20° C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCI (pH 7.6), 10 mM MgCI 2 , 5 mM DTT 及适量的NaCl 或KCI (视酶的具体要求而定)。
20%的PAGE (7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2. 双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara 公司从1979 年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37 C进行同步酶切。
但BamH I在37 C下有时表现出star活性,常用30 C单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。

各种酶切位点的保护碱基引物设计必看酶切位点是指特定的序列,酶可以识别并在该位置切割DNA分子。
这些位点的特异性使得酶在分子生物学中广泛应用于DNA片段的定位和切割。
然而,在一些实验中,我们可能需要保护酶切位点周围的碱基,以免酶切,并且只在特定的位置引导酶切。
因此,保护碱基引物的设计对于实验的成功非常重要。
以下是保护碱基引物设计的一些建议。
首先,保护碱基引物的设计需要考虑引物的长度。
引物的长度通常为18到30个碱基,具体的长度需要根据实验的需求和酶切位点周围的序列特征来确定。
引物的长度应足够长,以确保引物和靶序列的特异性,但不应过长,以免引物形成二级结构或与非特异性位点结合。
其次,保护碱基引物的设计需要考虑引物的碱基组成。
在设计引物时,建议尽量避免引物中出现酶切位点周围的碱基序列,以防止酶的误切。
例如,如果我们希望保护酶切位点周围的AATTC序列,可以设计一个引物,其中没有AATTC序列。
同时,引物的碱基组成应尽量避免多聚核苷酸或含有GC碱基的片段,以防止引物之间的结合或引物与非特异靶序列的结合。
此外,保护碱基引物的设计需要考虑引物的特异性。
在设计引物时,建议使用特异性的引物序列,以确保引物只与目标酶切位点结合。
可以通过使用生物信息学工具,如BLAST,来验证引物的特异性。
引物的特异性还可以通过调整引物的长度和碱基组成来进一步提高。
最后,保护碱基引物的设计需要考虑引物的热力学性质。
引物的热力学性质包括引物的熔解温度(Tm值)和引物之间的配对。
引物的Tm值与引物的碱基组成、长度和引物与靶序列之间的碱基配对相关。
可以使用在线工具,如NEB的Tm计算器,来计算引物的Tm值,并对不同的引物进行比较。
此外,引物之间的配对可以通过设计引物的末端序列来调整,例如末端的碱基配对或非配对等。
总结起来,保护碱基引物的设计需要考虑引物的长度、碱基组成、特异性和热力学性质。
通过合理设计引物,可以保护酶切位点周围的碱基,并在特定位置引导酶切,为实验的成功提供有力的保障。

酶切位点保护碱基-PCR引物设计用于限制性内切酶酶切反应来源:easylabs 发布时间:2009-11-08 查看次数:12704本文给出了分子克隆中常用限制性内切酶的保护碱基序列,如AccI,A flIII,AscI,AvaI,BamHI,BglII,BssHII,BstEII,BstXI,ClaI,E coRI,HaeIII,HindIII,KpnI,MluI,NcoI,NdeI,NheI,NotI,N siI,PacI,PmeI,PstI,PvuI,SacI,SacII,SalI,ScaI,SmaI,S peI,SphI,StuI,XbaI,XhoI,XmaI,为什么要添加保护碱基?在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A2 60单位的寡核苷酸。
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments (oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同 buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。
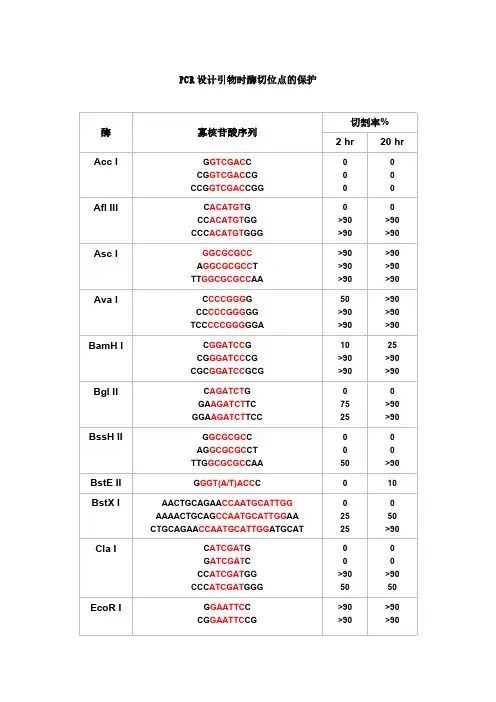
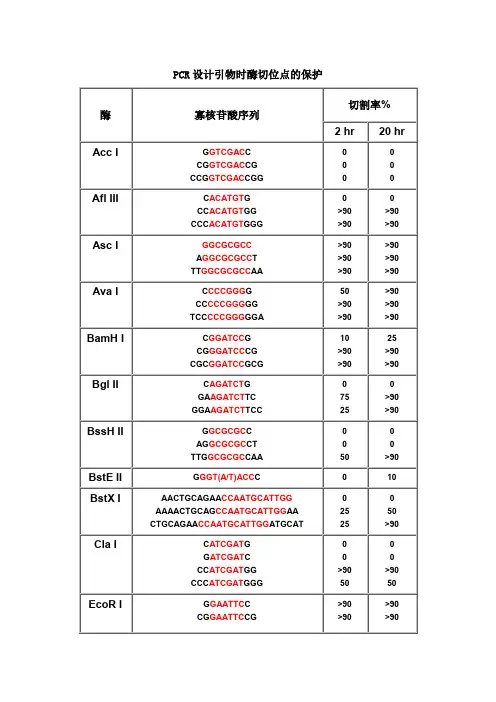
PCR设计引物时酶切位点的保护切割率%酶寡核苷酸序列2 hr20 hrAcc I G GTCGAC CCG GTCGAC CGCCG GTCGAC CGG 0Afl III C ACATGT GCC ACATGT GGCCC ACATGT GGG>90>90>90>90Asc I GGCGCGCCA GGCGCGCC TTT GGCGCGCC AA >90>90>90>90>90>90Ava I C CCCGGG GCC CCCGGG GGTCC CCCGGG GGA50>90>90>90>90>90BamH I C GGATCC GCG GGATCC CGCGC GGATCC GCG10>90>9025>90>90Bgl II C AGATCT GGA AGATCT TCGGA AGATCT TCC7525>90>90BssH II G GCGCGC CAG GCGCGC CTTTG GCGCGC CAA50>90BstE II G GGT(A/T)ACC C010BstX I AACTGCAGAA CCAATGCATTGGAAAACTGCAG CCAATGCATTGG AACTGCAGAA CCAATGCATTGG ATGCAT252550>90Cla I C ATCGAT GG ATCGAT CCC ATCGAT GGCCC ATCGAT GGG>9050>9050EcoR I G GAATTC CCG GAATTC CG >90>90>90>90CCG GAATTC CGG>90>90Hae III GG GGCC CCAGC GGCC GCTTTGC GGCC GCAA >90>90>90>90>90>90Hind III C AAGCTT GCC AAGCTT GGCCC AAGCTT GGG1075Kpn I G GGTACC CGG GGTACC CCCGG GGTACC CCG>90>90>90>90Mlu I G ACGCGT CCG ACGCGT CG2550Nco I C CCATGG GCATG CCATGG CATG5075Nde I C CATATG GCC CATATG GGCGC CATATG GCGGGGTTT CATATG AAACCCGGAATTC CATATG GAATTCCGGGAATTC CATATG GAATTCCC7575>90>90Nhe I G GCTAGC CCG GCTAGC CGCTA GCTAGC TAG10102550Not I TT GCGGCCGC AAATTT GCGGCCGC TTTAAAATAT GCGGCCGC TATAAAATAAGAAT GCGGCCGC TAAACTATAAGGAAAAAA GCGGCCGC AAAAGGAAAA10102525101090>90Nsi I TGC ATGCAT GCACCA ATGCAT TGGTTCTGCAGTT10>90>90>90Pac I TTAATTAAG TTAATTAA CCC TTAATTAA GG 025>90Pme I GTTTAAACG GTTTAAAC CGG GTTTAAAC CC 02550AGCTTT GTTTAAAC GGCGCGCCGG75>90Pst I G CTGCAG CTGCA CTGCAG TGCAAA CTGCAG AACCAATGCATTGGAAAA CTGCAG CCAATGCATTGGAACTGCAG AACCAATGCATTGGATGCAT10>90>9010>90>90Pvu I C CGATCG GAT CGATCG ATTCG CGATCG CGA102510Sac I C GAGCTC G1010Sac II G CCGCGG CTCC CCGCGG GGA50>90Sal I GTCGAC GTCAAAAGGCCATAGCGGCCGC GC GTCGAC GTCTTGGCCATAGCGGCCGCGGACGC GTCGAC GTCGGCCATAGCGGCCGCGGAA10105075Sca I G AGTACT CAAA AGTACT TTT 10752575Sma I CCCGGGC CCCGGG GCC CCCGGG GGTCC CCCGGG GGA10>90101050>90Spe I G ACTAGT CGG ACTAGT CCCGG ACTAGT CCGCTAG ACTAGT CTAG 1010>90>905050Sph I G GCATGC CCAT GCATGC ATGACAT GCATGC ATGT102550Stu I A AGGCCT TGA AGGCCT TCAAA AGGCCT TTT >90>90>90>90>90>90Xba I C TCTAGA GGC TCTAGA GCTGC TCTAGA GCACTAG TCTAGA CTAG>907575>90>90>90Xho I C CTCGAG GCC CTCGAG GGCCG CTCGAG CGG10102575Xma I C CCCGGG GCC CCCGGG GGCCC CCCGGG GGGTCCC CCCGGG GGGA2550>9075>90>90注释:1.如果要加在序列的5’端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3’端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。

各种酶切位点的保护碱基引物设计必看酶切位点的保护碱基引物设计在分子生物学领域中起着至关重要的作用。
它们是研究者在酶切实验中必不可少的工具,用于保护酶切位点周围的碱基,以避免酶的切割作用。
本文将介绍保护碱基引物设计的一般原则和具体步骤,并探讨一些常见的问题和注意事项。
保护碱基引物设计的一般原则如下:1.引物长度:保护碱基引物的长度通常为15-25个碱基对。
2.引物序列:引物应根据酶切位点的序列设计。
为了确保引物的特异性,通常将酶切位点和其周围的碱基考虑在内。
3.引物组成:引物的核苷酸组成应考虑碱基的GC含量,以保持引物的稳定性。
通常,GC含量高于50%的引物更稳定。
4.引物末端修饰:引物的末端修饰可以提高引物与目标DNA的亲和性,并增加引物的稳定性。
常用的末端修饰包括磷酸化和胺基修饰等。
保护碱基引物设计的步骤如下:1.获取酶切位点序列:首先,需要获取目标DNA序列中待保护的酶切位点的序列。
2.引物设计:根据酶切位点的序列设计引物。
引物的长度通常为15-25个碱基对。
为了提高特异性,可以考虑在引物序列中加入一些限制性内切酶无法识别的碱基。
3.引物末端修饰:根据需要选择引物的末端修饰方式,例如磷酸化和胺基修饰等。
4.引物的合成:完成引物设计后,可以委托专业的生物科技公司进行引物的合成。
确保引物的纯度和质量。
在进行保护碱基引物设计时,还需注意一些常见的问题和注意事项:1.引物特异性:在设计引物时,要确保引物与目标DNA的序列具有高度特异性,以避免引物与非目标区域的杂交。
2.引物的稳定性:引物的稳定性对于酶切实验的成功至关重要。
在设计引物时,要尽量选择稳定的引物序列,例如具有较高GC含量的引物。
3.引物纯度和质量:为了保证引物的质量和稳定性,引物的合成必须由专业的生物科技公司进行。
确保引物的纯度高,无杂质。
4.引物的浓度和稀释:在使用引物进行酶切实验时,要合理确定引物的浓度和稀释倍数,以保证实验的成功。
总之,保护碱基引物设计是分子生物学研究中不可或缺的一部分。
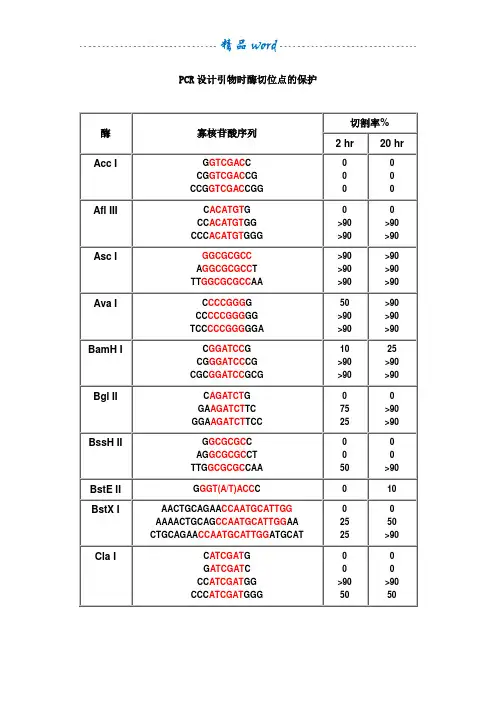
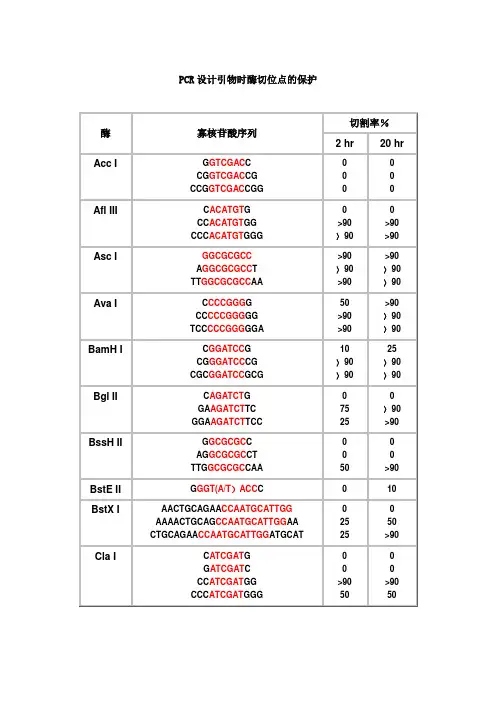
PCR设计引物时酶切位点的保护酶寡核苷酸序列切割率%2 hr20 hrAcc I G GTCGAC CCG GTCGAC CGCCG GTCGAC CGG 0Afl III C ACATGT GCC ACATGT GGCCC ACATGT GGG>90>90>90>90Asc I GGCGCGCCA GGCGCGCC TTT GGCGCGCC AA >90>90>90>90>90>90Ava I C CCCGGG GCC CCCGGG GGTCC CCCGGG GGA50>90>90>90>90>90BamH I C GGATCC GCG GGATCC CGCGC GGATCC GCG10>90>9025>90>90Bgl II C AGATCT GGA AGATCT TCGGA AGATCT TCC7525>90>90BssH II G GCGCGC CAG GCGCGC CTTTG GCGCGC CAA50>90BstE II G GGT(A/T)ACC C010BstX I AACTGCAGAA CCAATGCATTGGAAAACTGCAG CCAATGCATTGG AACTGCAGAA CCAATGCATTGG ATGCAT252550>90Cla I C ATCGAT GG ATCGAT CCC ATCGAT GGCCC ATCGAT GGG>9050>9050EcoR I G GAATTC CCG GAATTC CGCCG GAATTC CGG >90>90>90>90>90>90Hae III GG GGCC CCAGC GGCC GCTTTGC GGCC GCAA >90>90>90>90>90>90Hind III C AAGCTT GCC AAGCTT GGCCC AAGCTT GGG1075Kpn I G GGTACC CGG GGTACC CCCGG GGTACC CCG>90>90>90>90Mlu I G ACGCGT CCG ACGCGT CG2550Nco I C CCATGG GCATG CCATGG CATG5075Nde I C CATATG GCC CATATG GGCGC CATATG GCGGGGTTT CATATG AAACCCGGAATTC CATATG GAATTCCGGGAATTC CATATG GAATTCCC7575>90>90Nhe I G GCTAGC CCG GCTAGC CGCTA GCTAGC TAG10102550Not I TT GCGGCCGC AAATTT GCGGCCGC TTTAAAATAT GCGGCCGC TATAAAATAAGAAT GCGGCCGC TAAACTATAAGGAAAAAA GCGGCCGC AAAAGGAAAA10102525101090>90Nsi I TGC ATGCAT GCACCA ATGCAT TGGTTCTGCAGTT10>90>90>90Pac I TTAATTAAG TTAATTAA CCC TTAATTAA GG 025>90Pme I GTTTAAACG GTTTAAAC CGG GTTTAAAC CCAGCTTT GTTTAAAC GGCGCGCCGG752550>90Pst I G CTGCAG CTGCA CTGCAG TGCAAA CTGCAG AACCAATGCATTGGAAAA CTGCAG CCAATGCATTGGAACTGCAG AACCAATGCATTGGATGCAT10>90>9010>90>90Pvu I C CGATCG GAT CGATCG ATTCG CGATCG CGA102510Sac I C GAGCTC G1010Sac II G CCGCGG CTCC CCGCGG GGA50>90Sal I GTCGAC GTCAAAAGGCCATAGCGGCCGC GC GTCGAC GTCTTGGCCATAGCGGCCGCGGACGC GTCGAC GTCGGCCATAGCGGCCGCGGAA10105075Sca I G AGTACT CAAA AGTACT TTT 10752575Sma I CCCGGGC CCCGGG GCC CCCGGG GGTCC CCCGGG GGA10>90101050>90Spe I G ACTAGT CGG ACTAGT CCCGG ACTAGT CCGCTAG ACTAGT CTAG 1010>90>905050Sph I G GCATGC CCAT GCATGC ATGACAT GCATGC ATGT102550Stu I A AGGCCT TGA AGGCCT TCAAA AGGCCT TTT >90>90>90>90>90>90Xba IC TCTAGA GGC TCTAGA GCTGC TCTAGA GCACTAG TCTAGA CTAG>907575>90>90>90Xho I C CTCGAG GCC CTCGAG GGCCG CTCGAG CGG10102575Xma I C CCCGGG GCC CCCGGG GGCCC CCCGGG GGGTCCC CCCGGG GGGA2550>9075>90>90注释:1.如果要加在序列的5’端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3’端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。

本文给出了分子克隆中常用限制性内切酶的保护碱基序列,如AccI,AflIII,AscI,AvaI,BamHI,BglII,BssHII,BstEII,BstXI,ClaI,EcoRI,HaeIII,HindIII,KpnI,MluI,NcoI,NdeI,NheI,NotI,NsiI,PacI,PmeI,PstI,PvuI,SacI,SacII,SalI,ScaI,SmaI,SpeI,SphI,StuI,XbaI,XhoI,XmaI,为什么要添加保护碱基?在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
该如何添加保护碱基?添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A26单位的寡核苷酸。
取1µg已标记了的寡核苷酸与20单位的内切酶,在2 00°C条件下分别反应2小时和20小时。
各种酶切位点的保护碱基酶切位点是指酶在DNA或RNA分子中特定的位置识别并切割的区域。
在分子生物学研究中,酶切位点的保护碱基是进行引物设计的重要参考依据之一、本文将介绍各种酶切位点的保护碱基以及在引物设计中的应用。
1.核酸酶A切割位点保护碱基核酸酶A(RNase A)是一种特定的核酸酶,能够将单链RNA切割为5'-磷酸核酸和3'-核磷酸。
核酸酶A识别和切割位点的保护碱基主要为对尿嘧啶核苷酸(Uridine,U)和鸟嘌呤核苷酸(Adenine,A)。
具体来说,核酸酶A主要作用于RNA链上U和A的周围碱基,特别是位于U或A的下一个碱基。
在引物设计中,需要考虑核酸酶A切割位点的保护碱基,以避免产生无法预测的酶切割产物。
特别是在设计RNA引物时,需要避免在酶切位点附近出现U和A的保护碱基。
2.核酸酶T1切割位点保护碱基核酸酶T1(RNase T1)是一种特定的核酸酶,能够识别并切割由磷酸鸟苷(Guanosine,G)形成的RNA链。
核酸酶T1在RNA链上识别和切割位点的保护碱基为G。
具体来说,核酸酶T1主要作用于G的下一个碱基。
在引物设计中,需要考虑核酸酶T1切割位点的保护碱基,以避免产生无法预测的酶切割产物。
特别是在设计RNA引物时,需要避免在酶切位点附近出现G的保护碱基。
3.限制性内切酶切割位点保护碱基限制性内切酶是一类广泛应用于DNA分子生物学研究的酶,其识别和切割DNA分子中的特定序列。
限制性内切酶识别和切割位点的保护碱基由酶自身的特异性决定。
每一种限制性内切酶都有其特定的酶切位点保护碱基要求。
一般来说,酶切位点的保护碱基主要存在于酶切位点的上下游碱基中。
在引物设计中,需要考虑限制性内切酶切割位点的保护碱基,以避免引物和酶切位点之间存在相互作用而导致切割不完全或无法切割的情况。
总而言之,在引物设计中,需要考虑各种酶切位点的保护碱基,以提高引物的特异性和稳定性。
根据不同酶的切割特点和要求,设计合适的引物序列可以避免酶切位点的保护碱基产生干扰,保证实验结果的准确性。
PCR设计引物时酶切位点的保护切割率%酶寡核苷酸序列2 hr20 hrAcc I G GTCGAC CCG GTCGAC CGCCG GTCGAC CGG 0Afl III C ACATGT GCC ACATGT GGCCC ACATGT GGG>90>90>90>90Asc I GGCGCGCCA GGCGCGCC TTT GGCGCGCC AA >90>90>90>90>90>90Ava I C CCCGGG GCC CCCGGG GGTCC CCCGGG GGA50>90>90>90>90>90BamH I C GGATCC GCG GGATCC CGCGC GGATCC GCG10>90>9025>90>90Bgl II C AGATCT GGA AGATCT TCGGA AGATCT TCC7525>90>90BssH II G GCGCGC CAG GCGCGC CTTTG GCGCGC CAA50>90BstE II G GGT(A/T)ACC C010BstX I AACTGCAGAA CCAATGCATTGGAAAACTGCAG CCAATGCATTGG AACTGCAGAA CCAATGCATTGG ATGCAT252550>90Cla I C ATCGAT GG ATCGAT CCC ATCGAT GGCCC ATCGAT GGG>9050>9050EcoR I G GAATTC CCG GAATTC CG >90>90>90>90CCG GAATTC CGG>90>90Hae III GG GGCC CCAGC GGCC GCTTTGC GGCC GCAA >90>90>90>90>90>90Hind III C AAGCTT GCC AAGCTT GGCCC AAGCTT GGG1075Kpn I G GGTACC CGG GGTACC CCCGG GGTACC CCG>90>90>90>90Mlu I G ACGCGT CCG ACGCGT CG2550Nco I C CCATGG GCATG CCATGG CATG5075Nde I C CATATG GCC CATATG GGCGC CATATG GCGGGGTTT CATATG AAACCCGGAATTC CATATG GAATTCCGGGAATTC CATATG GAATTCCC7575>90>90Nhe I G GCTAGC CCG GCTAGC CGCTA GCTAGC TAG10102550Not I TT GCGGCCGC AAATTT GCGGCCGC TTTAAAATAT GCGGCCGC TATAAAATAAGAAT GCGGCCGC TAAACTATAAGGAAAAAA GCGGCCGC AAAAGGAAAA10102525101090>90Nsi I TGC ATGCAT GCACCA ATGCAT TGGTTCTGCAGTT10>90>90>90Pac I TTAATTAAG TTAATTAA CCC TTAATTAA GG 025>90Pme I GTTTAAACG GTTTAAAC CGG GTTTAAAC CC 02550AGCTTT GTTTAAAC GGCGCGCCGG75>90Pst I G CTGCAG CTGCA CTGCAG TGCAAA CTGCAG AACCAATGCATTGGAAAA CTGCAG CCAATGCATTGGAACTGCAG AACCAATGCATTGGATGCAT10>90>9010>90>90Pvu I C CGATCG GAT CGATCG ATTCG CGATCG CGA102510Sac I C GAGCTC G1010Sac II G CCGCGG CTCC CCGCGG GGA50>90Sal I GTCGAC GTCAAAAGGCCATAGCGGCCGC GC GTCGAC GTCTTGGCCATAGCGGCCGCGGACGC GTCGAC GTCGGCCATAGCGGCCGCGGAA10105075Sca I G AGTACT CAAA AGTACT TTT 10752575Sma I CCCGGGC CCCGGG GCC CCCGGG GGTCC CCCGGG GGA10>90101050>90Spe I G ACTAGT CGG ACTAGT CCCGG ACTAGT CCGCTAG ACTAGT CTAG 1010>90>905050Sph I G GCATGC CCAT GCATGC ATGACAT GCATGC ATGT102550Stu I A AGGCCT TGA AGGCCT TCAAA AGGCCT TTT >90>90>90>90>90>90Xba I C TCTAGA GGC TCTAGA GCTGC TCTAGA GCACTAG TCTAGA CTAG>907575>90>90>90Xho I C CTCGAG GCC CTCGAG GGCCG CTCGAG CGG10102575Xma I C CCCGGG GCC CCCGGG GGCCC CCCGGG GGGTCCC CCCGGG GGGA2550>9075>90>90注释:1.如果要加在序列的5’端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3’端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
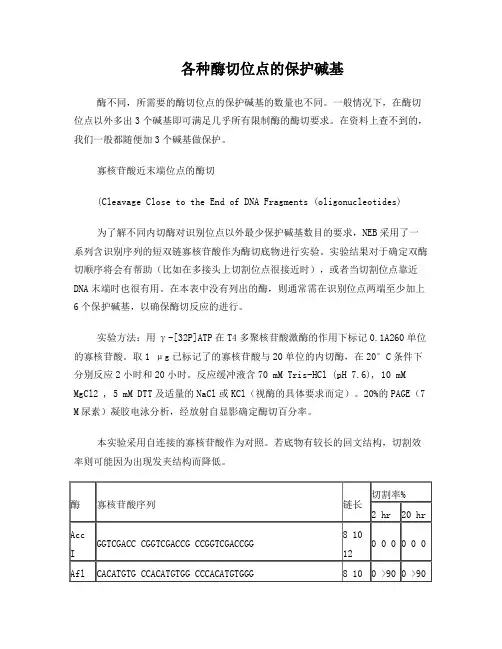
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切
(Cleavage Close to the End of DNA Fragments
(oligonucleotides)
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题
参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积
累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。
3.酶切底物DNA,切不开
1)底物DNA上没有相应的限制酶识别位点,或酶切位点被甲基化。
2)PCR引物的酶切位点前没有保护碱基或引物合成有误,致使没有正确的酶切位点存在。
PCR产物酶切前尽量进行精制以更换buffer。
由于PCR产物中带入的其它物质,会影响酶切,据报道,通常PCR产物的添加量占总反应体积25%以下没有问题。
3)酶切条件的确认,包括反应温度和反应体系等。
同样的DNA,同样量,用不同的限制酶切情况可能不同,由于DNA的空间结构造成的。
同样的DNA,不同的反应体系,酶切效果也可能不同,由于一些空间因素或不可测因素造成的。
4)公司出售的限制酶都是液体状态,都是根据最佳反应体系配制了浓度,不可以再用buffer稀释,因为酶浓度和活性之间不呈直线对应关系,酶浓度越稀,相对活性越低,并且越不稳定,有时便会出现底物DNA不能被切断的现象。
不同公司的酶和buffer不要交叉使用,否则可能会影响酶切效果。
5)酶的识别位点上的碱基被甲基化。
可以选用不受甲基化影响的同裂酶,
或将质粒DNA转入甲基化酶欠损的宿主菌中,重新制备DNA,也可以使用PCR的方法对DNA进行扩增,再做酶切。
常用的有XbaI容易受甲基化影响,通常选用GM33做宿主菌转化。
6)底物不纯,含有限制酶阻害物质,影响酶切作用,需要重新纯化DNA。
一般做乙醇沉淀纯化即可。
如果质粒中含盐或酚等,都会影响酶切效果。
4.DNA经酶切后,电泳无带或出现smear现象
DNA或酶切试剂中混有DNase,在一定的温度或缓冲液的作用下,激发DNase的作用,将DNA降解。
可以用DNA上的其他酶,也可以用此酶切其他DNA来检查问题所在。
如果质粒酶切出现此情况,首先将质粒做乙醇沉淀再酶切。
5.甲基化酶
M.Alu I,? M.Bam H I,? M.RcoR I不属于CG甲基化,dam甲基化,也不属于dcm甲基化。
甲基化酶作用后,DNA不能被相应的酶切断。