供注射用辅料XXXX版药典与各国药典质量标准对比表
- 格式:pdf
- 大小:8.41 MB
- 文档页数:44

分析方法预验证方案方案名称:XXXX有关物质测定方法预验证方案方案编号:版本号:01版起草人/日期:审核人/日期:审核人/日期:批准人/日期:目录1.概述 (1)2.分析方法 (1)2.1试剂 (1)2.2对照品 (1)2.3样品 (1)2.4仪器 (1)2.5色谱条件 (2)2.6溶液的配制 (2)2.7测定方法 (3)2.8计算 (3)3.方法验证 (3)3.1验证项目、验证方法及接受标准 (3)3.2系统适用性 (5)3.3专属性 (7)3.4精密度 (11)3.5线性 (12)3.6 定量限及检测限 (15)3.7准确度 (16)3.8溶液稳定性 (18)4.版本历史 (20)1.概述本品为xxxx注射液,规格为xxxx。
本品各国药典均未收载,本方法是参照欧洲药典(EP10.4)、美国药典(USP43)、中国药典2020年版收载的xxxx原料质量标准及英国药典(BP2020)收载的xxxx注射液质量标准及xxxx注射液进口注射标准(I物质A)项下的有关物质方法,采用中国药典2020年版四部通则0512高效液相色谱法,建立了本品的有关物质方法。
本预验证实验是为了确保该分析方法能够适用于当前本品有关物质的测定。
验证参数包括:系统适用性、专属性、重复性、精密度、线性、定量限(LOQ)、检测限(LOD)、准确度和溶液稳定性。
2.分析方法2.1试剂2.2对照品2.3样品2.4仪器2.5色谱条件色谱柱:C18,250×4.6mm,5μm或效能相当的色谱柱;检测波长:XXXnm;柱温:40℃;流速:1.0ml/min;进样体积:20μl;流动相A:0.2%磷酸氢二胺(pH7.5),流动相B:甲醇-异丙醇(87:13)。
梯度洗脱条件:时间(min)流动相A(%)流动相B(%)X X XX X XX X XX X XX X X2.6溶液的配制2.6.1 流动相流动相A:取磷酸氢二胺2g,加水1000ml溶解,用磷酸调pH值至7.5,过滤,取滤液,即得;流动相B:量取甲醇1000ml,超声,即得。

目录3.2.P.1 剂型及产品组成 (3)3.2.P.1.1剂型和产品组成 (3)3.2.P.1.2专用溶剂 (3)3.2.P.1.3包装 (3)3.2.P.2 产品开发 (3)3.2.P.2.1 处方组成 (4)3.2.P.2.1.1 原料药 (4)3.2.P.2.1.2 辅料 (7)3.2.P.2.2 制剂研究 (7)3.2.P.2.2.1 处方开发过程 (8)3.2.P.2.2.2 制剂相关特性 (14)3.2.P.2.3 生产工艺的开发 (16)3.2.P.2.3.1 小试工艺研究 (16)3.2.P.2.3.2 中试及验证生产工艺研究 (18)3.2.P.2.3.3 工艺变更情况汇总 (19)3.2.P.2.3.4代表性批次汇总 (19)3.2.P.2.4 包装材料/容器 (21)3.2.P.2.4.1 包材类型、来源及相关证明文件 (21)3.2.P.2.4.2 包材的选择依据 (21)3.2.P.2.4.3 包材选择的支持性研究 (22)3.2.P.2.5 相容性 (22)3.2.P.3 生产 (23)3.2.P.3.1 生产商 (23)3.2.P.3.2 批处方 (23)3.2.P.3.3 生产工艺和工艺控制 (23)3.2.P.3.3.1 工艺流程图 (23)3.2.P.3.3.2 工艺描述 (26)3.2.P.3.3.3 主要的生产设备 (26)3.2.P.3.3.4 拟定的大生产规模 (26)3.2.P.3.4关键步骤和中间体的控制 (26)3.2.P.3.4.1 关键步骤和工艺参数 (27)3.2.P.3.4.2 中间体的控制 (27)3.2.P.3.5 工艺验证和评价 (30)3.2.P.3.5.1 冻干粉针剂培养基模拟灌装验证 (30)3.2.P.3.5.2 除菌过滤系统验证 (30)3.2.P.3.5.3 内包材密封完整性验证 ........................................................ 错误!未定义书签。

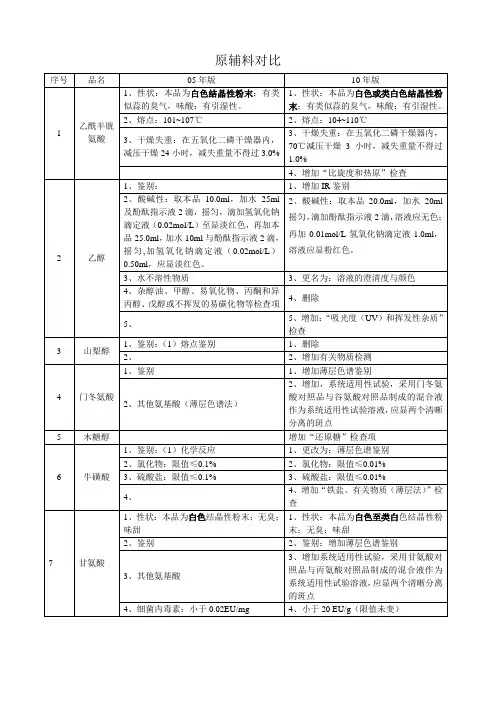
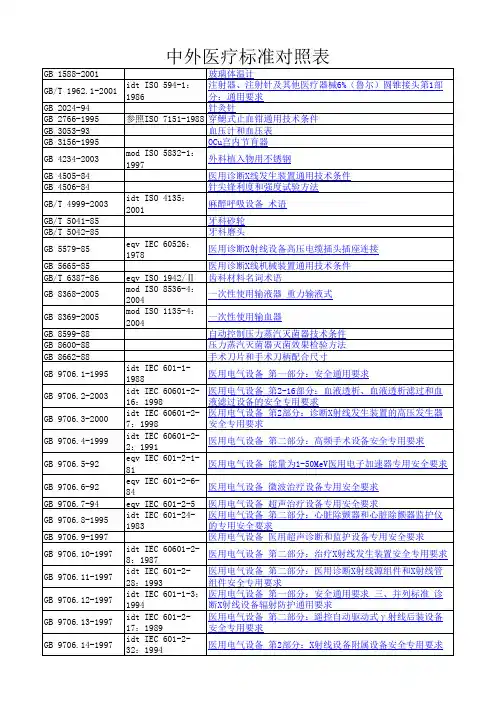
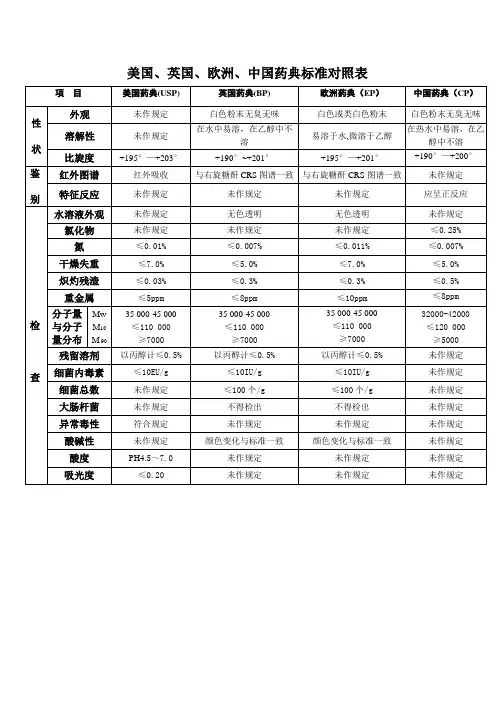
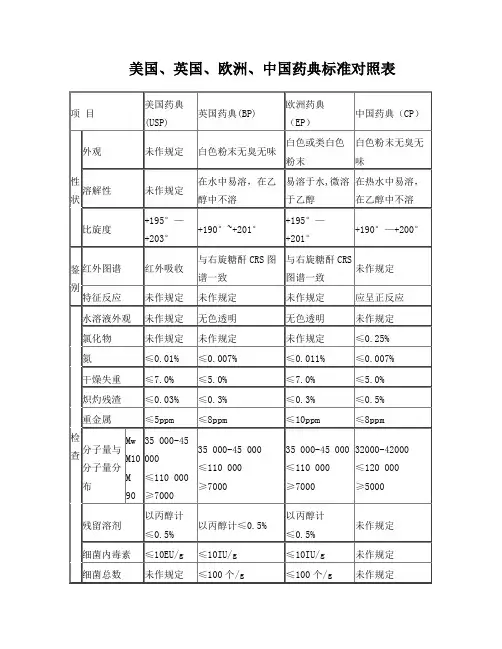
美国、英国、欧洲、中国药典标准对照表项目美国药典(USP) 英国药典(BP) 欧洲药典(EP)中国药典(CP)性状外观未作规定白色粉末无臭无味白色或类白色粉末白色粉末无臭无味溶解性未作规定在水中易溶,在乙醇中不溶易溶于水,微溶于乙醇在热水中易溶,在乙醇中不溶比旋度+195°—+203°+190°~+201°+195°—+201°+190°—+200°鉴别红外图谱红外吸收与右旋糖酐CRS图谱一致与右旋糖酐CRS图谱一致未作规定特征反应未作规定未作规定未作规定应呈正反应检查水溶液外观未作规定无色透明无色透明未作规定氯化物未作规定未作规定未作规定≤0.25%氮≤0.01%≤0.007%≤0.011%≤0.007%干燥失重≤7.0%≤5.0%≤7.0%≤5.0%炽灼残渣≤0.03%≤0.3%≤0.3%≤0.5%重金属≤5ppm≤8ppm≤10ppm≤8ppm分子量与分子量分布MwM10M 9035 000-45 000≤110 000≥700035 000-45 000≤110 000≥700035 000-45 000≤110 000≥700032000-42000≤120 000≥5000残留溶剂以丙醇计≤0.5%以丙醇计≤0.5%以丙醇计≤0.5%未作规定细菌内毒素≤10EU/g≤10IU/g≤10IU/g未作规定细菌总数未作规定≤100个/g≤100个/g未作规定大肠杆菌未作规定不得检出不得检出未作规定异常毒性符合规定未作规定未作规定未作规定酸碱性未作规定颜色变化与标准一致颜色变化与标准一致未作规定酸度PH4.5~7.0未作规定未作规定未作规定吸光度≤0.20未作规定未作规定未作规定。



各国药典比较ChP、USP、Ph.Eur.中药/天然药物质量标准比较及评述—以芦荟为例by14211第一部分中美欧药典简介1.中国药典《中华人民共和国药典》,简称《中国药典》。
是由国家药典委员会(原名卫生部药典委员会成立于1950年),根据《中华人民共和国药品管理法》的规定,负责组织编纂《中华人民共和国药典》及制定、修订国家药品标准,是法定的国家药品标准。
由国家食品药品监督管理部门批准颁布实施。
《中华人民共和国药典》(简称《中国药典》)2010年版,分一部、二部和三部,收载品种总计4567种,其中新增1386种。
药典一部收载药材和饮片、植物油脂和提取物、成方制剂和单味制剂等,品种共计2165种,其中新增1019种(包括439个饮片标准)、修订634种;药典二部收载化学药品、抗生素、生化药品、放射性药品以及药用辅料等,品种共计2271种,其中新增330种、修订1500种;药典三部收载生物制品,品种共计131种,其中新增37种、修订94种。
2010版药典收载的附录亦有变化,其中药典一部新增14个、修订47个;药典二部新增15个、修订69个;药典三部新增18个、修订39个。
一、二、三部共同采用的附录分别在各部中予以收载,并尽可能做到统一协调、求同存异。
中国药典包括凡例、正文及附录,是药品研制、生产、经营、使用和监督管理等均应遵循的法定依据。
所有国家药品标准应当符合中国药典凡例及附录的相关要求。
作为我国保证药品质量的法典,本版药典在保持科学性、先进性、规范性和权威性的基础上,着力解决制约药品质量与安全的突出问题,着力提高药品标准质量控制水平,充分借鉴了国际先进技术和经验,客观反映了中国当前医药工业、临床用药及检验技术的水平,必将在提高药品质量过程中起到积极而重要的作用,并将进一步扩大和提升我国药典在国际上的积极影响。
2.美国药典《美国药典/国家处方集》U.S. Pharmacopeia / National Formulary(简称USP/NF)。
目录3.2.P.1 剂型及产品组成 (3)3.2.P.1.1剂型和产品组成 (3)3.2.P.1.2专用溶剂 (3)3.2.P.1.3包装 (3)3.2.P.2 产品开发 (3)3.2.P.2.1 处方组成 (4)3.2.P.2.1.1 原料药 (4)3.2.P.2.1.2 辅料 (7)3.2.P.2.2 制剂研究 (7)3.2.P.2.2.1 处方开发过程 (8)3.2.P.2.2.2 制剂相关特性 (14)3.2.P.2.3 生产工艺的开发 (16)3.2.P.2.3.1 小试工艺研究 (16)3.2.P.2.3.2 中试及验证生产工艺研究 (18)3.2.P.2.3.3 工艺变更情况汇总 (19)3.2.P.2.3.4代表性批次汇总 (19)3.2.P.2.4 包装材料/容器 (21)3.2.P.2.4.1 包材类型、来源及相关证明文件 (21)3.2.P.2.4.2 包材的选择依据 (21)3.2.P.2.4.3 包材选择的支持性研究 (22)3.2.P.2.5 相容性 (22)3.2.P.3 生产 (23)3.2.P.3.1 生产商 (23)3.2.P.3.2 批处方 (23)3.2.P.3.3 生产工艺和工艺控制 (23)3.2.P.3.3.1 工艺流程图 (23)3.2.P.3.3.2 工艺描述 (26)3.2.P.3.3.3 主要的生产设备 (26)3.2.P.3.3.4 拟定的大生产规模 (26)3.2.P.3.4关键步骤和中间体的控制 (26)3.2.P.3.4.1 关键步骤和工艺参数 (27)3.2.P.3.4.2 中间体的控制 (27)3.2.P.3.5 工艺验证和评价 (30)3.2.P.3.5.1 冻干粉针剂培养基模拟灌装验证 (30)3.2.P.3.5.2 除菌过滤系统验证 (30)3.2.P.3.5.3 内包材密封完整性验证 ........................................................ 错误!未定义书签。
CTD格式模板(制剂)资料CTD格式申报资料撰写格式(制剂)3.2.P.1 剂型及产品组成3.2.P.2 产品开发3.2.P.2.1 处⽅组成3.2.P.2.1.1 原料药3.2.P.2.1.2 辅料3.2.P.2.2 制剂3.2.P.2.2.1 处⽅开发过程3.2.P.2.2.2 制剂相关特性3.2.P.2.3 ⽣产⼯艺的开发3.2.P.2.4 包装材料/容器3.2.P.2.5 相容性3.2.P.3 ⽣产3.2.P.3.1 ⽣产商3.2.P.3.2 批处⽅3.2.P.3.3 ⽣产⼯艺和⼯艺控制3.2.P.3.4 关键步骤和中间体的控制3.2.P.3.5 ⼯艺验证和评价3.2.P.4 原辅料的控制3.2.P.5 制剂的质量控制3.2.P.5.1 质量标准3.2.P.5.2 分析⽅法3.2.P.5.3 分析⽅法的验证3.2.P.5.4 批检验报告3.2.P.5.5 杂质分析3.2.P.5.6 质量标准制定依据3.2.P.6 对照品3.2.P.7 稳定性3.2.P.7.1 稳定性总结3.2.P.7.2 上市后的稳定性研究⽅案及承诺3.2.P.7.3 稳定性数据CTD格式8号申报资料主要研究信息(药学部分:制剂)3.2.P.1 剂型选择依据及产品组成(1)本品为普通⽚剂,规格为:10mg每⽚XXXXX⽚的处⽅组成见表3.2.P.1。
附表3.2.P.1 XXXXX⽚药物组成表成份⽤量是否过量加⼊作⽤执⾏标准(2)本品在制备过程中没有使⽤专⽤的溶剂。
(3)XXXXX⽚的内包装材料为铝塑板,有避光和防潮的作⽤,与XXXXX 的内包装材质相同。
内包材的⽣产⼚家具有内包材注册证,执⾏国家标准。
3.2.P.2 产品开发XXXXX是HMG-CoA还原酶的⼀选择性、竞争性抑制剂,可以显著降低胆固醇⽔平,并降低⼼肌梗死或脑卒中的发病危险。
临床试验已经证实XXXXX降低胆固醇的临床疗效明显优于其它汀类药物,对原发性⾼胆固醇⾎症、包括家族性⾼胆固醇⾎症或混合型⾼脂⾎症患者以及纯合⼦家族性⾼胆固醇⾎症者有明显疗效。
各国药用辅料标准对比手册
(数字版)
V1.0
用户手册
9附录
9.1各国药用辅料标准收载品种索引
38
2010 2015 38 8.5 16
2010 2015
2010 2015 38 8.5 16
38
2015 38 8.5 16
38
38 8.5
2015 38 8.5
38 8.5
2010 2015 38 8.5
38
2010 2015 38 8.5
2010 2015
38
38
2015 38
38
2015 38
2010 2015 38 8.5
2010 2015
1.本书在编译时维持了各国药典药用辅料标准的体例和单位书写方式。
2.部分品种在2010 年版药典二部作为原料药品种收载,2015 年版药典
四部增订了相应药用辅料品种。
3.收录的《中国药典》2010 年版药用辅料标准所采用检定方法的“附
录”系指原《中国药典》2010 年版二部收载附录。
9.2售后及技术支持
国家药典委员会
根据《中华人民共和国药品管理法》规定,国家药典委员会负责组织编纂
《中华人民共和国药典》及制定、修订国家药品标准,是法定的国家药品
标准工作专业管理机构。
科迈恩(北京)科技有限公司
作为复杂体系先进质量控制解决方案的提出者,科迈恩科技致力于为药
品、医疗、食品、仪器分析等领域提供专业级大数据解决方案。
附件:丙二醇(供注射用)Bing’erchun(Gongzhusheyong)Propylene Glycol(For Injection)OHOHH3CC3H8O276.09 本品为1,2-丙二醇,含C3H8O2不得少于99.0% 。
【性状】本品为无色澄清的黏稠液体;无臭;有引湿性。
本品与水、乙醇或三氯甲烷能任意混溶。
相对密度本品的相对密度(通则0601)在25°C 时应为1.035~1.037。
折光率本品的折光率(通则0622),应为1.431~1.433。
【鉴别】(1)在含量测定项下记录的色谱图中,供试品溶液各主峰的保留时间应与对照品溶液相应主峰的保留时间一致。
(2)本品的红外光吸收图谱应与对照的图谱(光谱集706图)一致(通则0402)。
【检查】酸度取本品10.0ml,加新沸放冷的水50ml溶解后,加溴麝香草酚蓝指示液 3 滴,用氢氧化钠滴定液(0.01mol/L)滴定至溶液显蓝色,消耗氢氧化钠滴定液(0.01mol/L)的体积不得过0.5ml。
氯化物取本品1.0ml,依法检查(通则0801),与标准氯化钠溶液7.0m l制成的对照液比较,不得更浓(0.007%)。
硫酸盐取本品5.0ml,依法检查(通则0802) ,与标准硫酸钾溶液 3.0m l制成的对照液比较,不得更浓(0.006%)。
有关物质取本品适量,精密称定,用无水乙醇稀释制成每1ml中含丙二醇0.5g的溶液,作为供试品溶液;另精密称取一缩二乙二醇(二甘醇)、一缩二丙二醇、二缩三丙二醇和环氧丙烷对照品,用无水乙醇稀释制成每1ml中各约含5μg、500μg、150μg和5μg的混合溶液,作为对照品溶液。
照气相色谱法(通则0521)试验。
以聚乙二醇20M为固定液的毛细管柱为色谱柱,起始温度为80℃,维持3 分钟,以每分钟15℃的速率升温至220℃,维持4分钟,进样口温度230℃,检测器温度250℃,各组分峰的分离度应符合要求。
《中国药典》、《美国药典》和《英国药典》注射用氢化可的松琥珀酸钠质量控制标准与方法的比较张莹【摘要】本文讨论2010年版《中国药典》注射用氢化可的松琥珀酸钠新增内容,并与《美国药典》(35版)和《英国药典》(2010年版)比较,2010年版《中国药典》注射用氢化可的松琥珀酸钠较2005年版《中国药典》增加了红外鉴别及有关物质检查,对杂质和有害物质的控制进一步加强,以保证药品的质量.【期刊名称】《天津药学》【年(卷),期】2012(024)006【总页数】4页(P6-8,35)【关键词】药典;氢化可的松琥珀酸钠;标准;比较【作者】张莹【作者单位】天津市药品检验所,天津300070【正文语种】中文【中图分类】R927.11氢化可的松琥珀酸钠是肾上腺皮质激素类药,具有抗炎、抗过敏和抑制免疫等多种药理作用。
目前,注射用氢化可的松琥珀酸钠收载于《中国药典》2010年版[1]、《美国药典》35 版[2]和《英国药典》2010年版[3]中。
通过对三国药典的比较,对于进一步完善和提高我国药品质量标准具有指导意义,并指出下一步标准提高的方向。
1 2010年版《中国药典》与2005年版《中国药典》的比较1.1 性状外观描述同《中国药典》2005年版二部原质量标准[1],未做修订。
1.2 鉴别化学反应、高效液相色谱法及钠盐鉴别反应同《中国药典》2005年二部原质量标准,方法未做修订。
《中国药典》2010年版增加了红外光吸收图谱鉴别[2],标准规定样品的红外吸收图谱应与对照的图谱(光谱集994图)一致。
1.3 检查1.3.1 碱度、溶液的颜色、干燥失重、异常毒性、无菌及其他项目检查均同《中国药典》2005年二部原质量标准,方法未做修订。
1.3.2 有关物质《中国药典》2005年二部原质量标准未制订此项检查,只控制游离氢化可的松的限度。
由于氢化可的松琥珀酸钠在合成中主要产物为21-氢化可的松琥珀酸钠,但也不同程度地存在17-氢化可的松琥珀酸钠和游离氢化可的松以及未知的其他有关物质,为此,《中国药典》2010年版增加了有关物质检查,色谱条件采用含量测定项下的色谱条件,限度规定按外标法以峰面积计算游离氢化可的松不得过标示量的3.0%,其他单个杂质峰面积不得大于对照溶液主峰面积的1/3(1.0%),其他杂质峰面积的和不得大于对照溶液主峰面积(3.0%),该项目的增订可进一步控制药品的质量。
注射用水质量标准对比表
项目中国药典(2010版)欧洲药典6.0 美国药典usp31
来源本品为纯化水经蒸馏所
得本品为符合法定标准的
饮用水或纯化水经适当
方法蒸馏所得
由符合美国环保署、欧
共体、日本法定要求或
WHO饮用水指南的饮
用水为原水,经蒸馏或
蒸馏法去除化学物质及
微生物水平相当或更优
的纯化工艺制得
性质无色澄清液体;无臭,
无味无色澄清液体;无臭,
无味
N/A
PH值 5.0-7.0 N/A N/A
氨≤0.2μg/ml N/A N/A
硝酸盐≤0.06μg/ml≤0.2μg/ml N/A
亚硝酸盐≤0.02μg/ml N/A N/A
重金属≤0.1μg/ml≤0.1μg/ml N/A
不挥发物不得超过0.01mg/ml N/A N/A
铝盐N/A 生产渗析液时控制此项N/A
总有机碳≤0.5mg/l≤0.5mg/l(≤500ppb) ≤0.5mg/l(≤500ppb) 电导率应符合规定应符合规定应符合规定
(1.1μs/cm@20℃
1.3μs/cm@20℃)
细菌内毒素≤0.25EU/m ≤0.25EU/ml ≤0.25EU/m
微生物限度≤10CFU/100ml ≤10CFU/100ml ≤10CFU/100ml。