二类新药申报流程
- 格式:docx
- 大小:12.24 KB
- 文档页数:1


中药新药研发申报流程及相关申报材料说明
一、 中药新药的注册分类及说明
1.1 注册分类
中药新药注册按审批管理的要求分以下几类:
1. 未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。
2. 新发现的药材及其制剂。
3. 新的中药材代用品.
4. 药材新的药用部位及其制剂。
5. 未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。
6. 未在国内上市销售的中药、天然药物复方制剂。
7. 改变国内已上市销售中药、天然药物给药途径的制剂。
8. 改变国内已上市销售中药、天然药物剂型的制剂。
9. 仿制药。 1.2 说明
注册分类1—6的品种为新药,注册分类7、8按新药申请程序申报,注册分类9的品种为已有国家标准的药品。
二、 中药新药的研发及申报流程
中药新药的研发申报一般按以下程序进行:
选题立项——临床前研究—-临床研究——申报审批——正式生产,其中,新药临床前及临床研究的主要内容及注意事项分别列举如下:
2.1 新药的临床前研究
(一)主要内容:
新药的临床前研究主要包括制备工艺(中药制剂包括原药材的来源、加工及炮制)、理化性质、纯度、检验方法、处方筛选、剂型、稳定性、质量标准、药理、毒理、动物药代动力学等研究。新发现中药材还应包括来源、生态环境、栽培(养殖)技术、采收处理、加工炮制等研究。
(二)注意事项:
从事新药安全性研究的实验室应符合国家药品监督管理局《药品非临床研究质量管理规范》(GLP)的相应要求,实验动物应符合国家药品监督管理局的有关要求,以保证各项实验的科学性和实验结果的可靠性。
2.2 新药的临床研究
(一)主要内容: 新药的临床研究包括临床试验和生物等效性试验。
新药的临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。
Ⅰ期临床试验:初步的临床药理学及人体安全性评价试验。观察人体对于新药的耐受程度和药物代谢动力学,为制定给药方案提供依据。
Ⅱ期临床试验:随机盲法对照临床试验。对新药有效性及安全性作出初步评价,推荐临床给药剂量。

中药新药研发申报流程及相关申报材料说明
一、 中药新药的注册分类及说明
1.1 注册分类
中药新药注册按审批治理的要求分以下几类:
1. 未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。
2. 新发觉的药材及其制剂。
3. 新的中药材代用品。
4. 药材新的药用部位及其制剂。
5. 未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。
6. 未在国内上市销售的中药、天然药物复方制剂。
7. 改变国内已上市销售中药、天然药物给药途径的制剂。
8. 改变国内已上市销售中药、天然药物剂型的制剂。
9. 仿造药。
1.2 说明
注册分类1-6的品种为新药,注册分类7、8按新药申请程序申报,注册分类9的品种为已有国家标准的药品。
二、 中药新药的研发及申报流程
中药新药的研发申报一样按以下程序进行:
选题立项——临床前研究——临床研究——申报审批——正式生产,其中,新药临床前及临床研究的要紧内容及注意事项别离列举如下:
新药的临床前研究
(一)要紧内容:新药的临床前研究要紧包括制备工艺(中药制剂包括原药材的来源、加工及炮制)、理化性质、纯度、查验方式、处方挑选、剂型、稳固性、质量标准、药理、毒理、动物药代动力学等研究。新发觉中药材还应包括来源、生态环境、栽培(养殖)技术、采收处置、加工炮制等研究。
(二)注意事项:
从事新药平安性研究的实验室应符合国家药品监督治理局《药品非临床研究质量治理标准》(GLP)的相应要求,实验动物应符合国家药品监督治理局的有关要求,以保证各项实验的科学性和实验结果的靠得住性。
新药的临床研究
(一)要紧内容:
新药的临床研究包括临床实验和生物等效性实验。
新药的临床实验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。
Ⅰ期临床实验:初步的临床药理学及人体平安性评判实验。观看人体关于新药的耐受程度和药物代谢动力学,为制定给药方案提供依据。
Ⅱ期临床实验:随机盲法对照临床实验。对新药有效性及平安性作出初步评判,推荐临床给药剂量。

一、 中药新药的注册分类及说明
1.1 注册分类
中药新药注册按审批管理的要求分以下几类:
1. 未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。
2. 新发现的药材及其制剂。
3. 新的中药材代用品。
4. 药材新的药用部位及其制剂。
5. 未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。
6. 未在国内上市销售的中药、天然药物复方制剂。
7. 改变国内已上市销售中药、天然药物给药途径的制剂。
8. 改变国内已上市销售中药、天然药物剂型的制剂。
9. 仿制药。
1.2 说明
注册分类1-6的品种为新药,注册分类7、8按新药申请程序申报,注册分类9的品种为已有国家标准的药品。
二、 中药新药的研发及申报流程
中药新药的研发申报一般按以下程序进行:
选题立项——临床前研究——临床研究——申报审批——正式生产,其中,新药临床前及临床研究的主要内容及注意事项分别列举如下:
新药的临床前研究
(一)主要内容:
新药的临床前研究主要包括制备工艺(中药制剂包括原药材的来源、加工及炮制)、理化性质、纯度、检验方法、处方筛选、剂型、稳定性、质量标准、药理、毒理、动物药代动力学等研究。新发现中药材还应包括来源、生态环境、栽培(养殖)技术、采收处理、加工炮制等研究。
(二)注意事项:
从事新药安全性研究的实验室应符合国家药品监督管理局《药品非临床研究质量管理规范》(GLP)的相应要求,实验动物应符合国家药品监督管理局的有关要求,以保证各项实验的科学性和实验结果的可靠性。
新药的临床研究
(一)主要内容:
新药的临床研究包括临床试验和生物等效性试验。
新药的临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。 Ⅰ期临床试验:初步的临床药理学及人体安全性评价试验。观察人体对于新药的耐受程度和药物代谢动力学,为制定给药方案提供依据。
Ⅱ期临床试验:随机盲法对照临床试验。对新药有效性及安全性作出初步评价,推荐临床给药剂量。
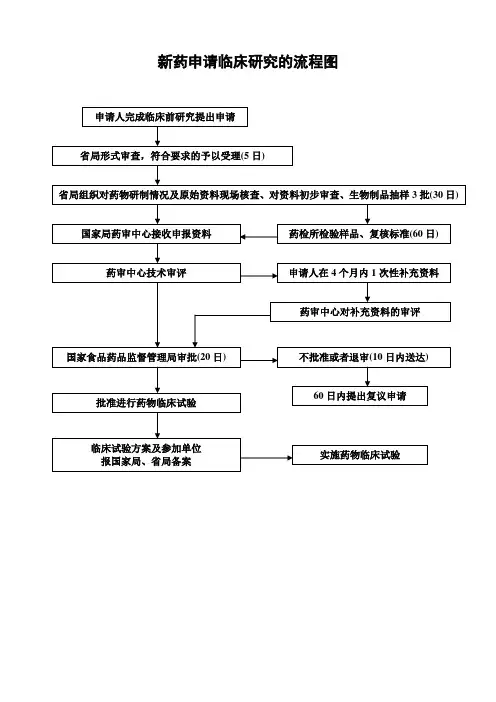
新药申请临床研究的流程图
申请人完成临床前研究提出申请
省局形式审查,符合要求的予以受理(5日)
省局组织对药物研制情况及原始资料现场核查、对资料初步审查、生物制品抽样3批(30日)
国家局药审中心接收申报资料 药检所检验样品、复核标准(60日)
药审中心技术审评 申请人在4个月内1次性补充资料
药审中心对补充资料的审评
国家食品药品监督管理局审批(20日) 不批准或者退审(10日内送达)
批准进行药物临床试验
临床试验方案及参加单位
报国家局、省局备案 实施药物临床试验 60日内提出复议申请
新药申请生产的审批流程图
申请人完成临床试验提出申请
省局形式审查,符合要求的予以受理(5日)
省局组织对临床试验情况及原始资料现场核查、申报资料初步审查、抽样3批(30日)
国家局药审中心接收申报资料 药检所检验样品、复核标准 (60日)
药审中心技术审评 申请人在4个月内1次性补充资料
药审中心对补充资料的审评
国家局审批(20日)
不批准或退审(10日内送达) 国家局药品认证中心
药品认证中心组织生产现场检查、抽样
药审中心形成综合意见
国家食品药品监督管理局审批(20日) 不批准或退审(10日内送达)
批准生产 60日内提出复议申请
60日内提出复议申请 仿制药的申报与审批流程
申请人提出申请
省局形式审查、组织对研制情况及原始资料现场核查、生产现场检查、抽样3批(35日)
国家局药审中心接收资料 药检所检验样品、复核标准(60日)
药审中心技术审评 申请人在4个月内1次性补充资料
药审中心对补充资料的审评
国家食品药品监督管理局审批(20日) 不批准或退审(10日内送达)
批准进行临床试验 批准生产
临床试验方案及参加单位
报国家局、省局备案
实施临床试验
申请人完成临床试验后向药审中心报送临床试验资料
药审中心技术审评
国家食品药品监督管理局审批(20日) 不批准或退审(10日内送达)