量子化学第五章分子轨道理论
- 格式:doc
- 大小:509.00 KB
- 文档页数:24





分子轨道理论1. 引言分子轨道理论是化学中的一种重要理论,它用量子力学的原理解释了分子的电子结构和化学性质。
本文将介绍分子轨道理论的基本概念、应用以及相关的计算方法。
2. 基本概念2.1 原子轨道在分子轨道理论中,首先要了解的是原子轨道。
原子轨道是描述单个原子中电子运动的波函数。
根据量子力学的原理,一个原子可以存在多个不同的原子轨道,每个原子轨道都对应着不同的能量状态。
2.2 分子轨道当两个或更多个原子靠近形成化学键时,原子轨道会互相重叠,形成新的分子轨道。
分子轨道描述的是电子在整个分子中的运动状态。
根据分子轨道理论,分子轨道可以分为两类:成键分子轨道和反键分子轨道。
成键分子轨道对应着电子的主要分布区域,而反键分子轨道则对应着电子分布相对较少的区域。
2.3 分子轨道能级分子轨道能级与原子轨道能级类似,分子轨道的能量随着轨道的能级增加而增加。
有时,分子轨道能级之间会有一定的能隙,这种能隙反映了分子稳定性的特征。
3. 分子轨道的应用分子轨道理论可以解释大量的化学现象和性质,下面列举了几个常见的应用:3.1 化学键的形成分子轨道理论提供了解释化学键产生的机制。
当两个原子靠近并形成化学键时,原子轨道会发生线性组合形成分子轨道。
通过分子轨道理论,我们可以理解不同类型的化学键(如共价键、离子键和金属键)是如何形成的以及其性质的差异。
3.2 分子轨道的能级顺序分子轨道理论还可以预测分子轨道的能级顺序,从而解释分子的化学性质。
能级较低的分子轨道通常具有较高的稳定性,从而决定了分子的化学反应性质。
3.3 分子光谱在分子光谱中,分子轨道理论被广泛应用。
分子轨道理论可以解释分子在吸收或发射光的过程中所发生的能级跃迁,从而解释不同光信号的产生和分子结构的变化。
4. 分子轨道的计算方法4.1 原子轨道模型著名的原子轨道计算方法包括Hartree-Fock方法和密度泛函理论。
这些方法通过求解原子的薛定谔方程,得到原子轨道及其能量。



分子轨道理论的基本概念分子轨道理论是描述分子内电子结构的理论基础,是理解和预测分子性质的重要工具。
它通过对分子中电子行为的定量描述,为我们提供了深入理解分子结构和化学性质的途径。
本文将介绍分子轨道理论的基本概念,包括分子轨道的形成、分子轨道能级、分子轨道的排布规律等内容。
分子轨道分子轨道是描述整个分子内所有电子运动状态的波函数。
在分子轨道理论中,通过线性组合原子轨道(Linear Combination of Atomic Orbitals, LCAO)方法,可以得到分子轨道波函数。
例如,两个氢原子相互结合形成氢气分子的过程中,每个原子的1s轨道可以线性组合形成一个成键分子轨道和一个反键分子轨道。
这种过程称为成键和反键形成。
通过这种方式形成的分子轨道波函数,可以用来描述氢气分子内电子的运动状态。
分子轨道能级根据量子力学原理,不同类型的分子轨道具有不同的能级。
一般来说,成键分子轨道的能级较低,反键分子轨道的能级较高。
在填充电子时,按照Pauli不相容原理和Hund规则,电子会依次填充到较低能级的成键分子轨道上,直到所有电子填充完毕。
这种填充顺序决定了分子的稳定性和化学性质。
分子轨道排布规律根据对称性和能量原理,我们可以确定不同类型分子轨道在空间中的排布规律。
以双原子分子为例,通过简单的组合对称性和量子力学计算,可以得到成键σ、反键σ、成键π和反键π四种主要类型的分子轨道。
每一种类型的分子轨道在空间中具有特定形状和取向,并且对应着不同的能级。
分子轨道理论在实践中的应用凭借其对化学键性质和反应活性等方面的深刻认识,分子轨道理论在近现代化学研究中扮演了重要角色。
它被广泛应用于有机合成设计、催化剂设计、光催化材料设计等领域。
例如,在有机合成设计中,我们可以通过对不同配体结构下电荷传递与空间排布特性进一步加深对反应机制及活性位点与其实际功能之间关联作用进一步了解。
结论总之,分子轨道理论为我们提供了揭示和预测化学现象背后原理的新视角,并且在许多实际应用中发挥着重要作用。

第五章分子轨道理论5.1 Hatree-Fock 方程Hatree-Fock 近似,也就是分子轨道近似,是量子化学中心之一,分子中的电子占据轨道,这是化学家头脑中很容易想到的。
首先,我们推导一下Hatree-Fock 方程。
由于绝大多数分子都是闭壳层的,因此我们都可以用单slater 行列式作为其波函数,即12N C f f f ψ=设我们有正交集i j ij f f δ= 则一、二阶约化密度矩阵为:'*'11111''111112''21212''112122(,)()()(,)(,)1(,;,)2(,)(,)i i ix x f x f x x x x x x x x x x x x x ρρρρρρ∧∧∧∧∧∧==∑改写一下(Dirac ):*'*'11122*'*'2122''1212()()()()12()()()()1[()()()()]2NNi i i i iiNNj j j j jjN i j i j i jj i i jf x f x f x f x f x f x f x f x f x f x f x f x f f f f ρ∧≠==-∑∑∑∑∑12(1)(1,2)1(1)[(1,2)(1,2)]2(1,2)(1,2)1[]2r r Ni i i j i j i j j i ii ji i i ii i i i Ni i i j i j i j j i iijE T h T g f h f f f g f f f f g f f f f g f f f f g f f E f h f f f g f f f f g f f ρρ∧∧∧∧≠=+=+--=+-∑∑∑∑因为i=j 时,=0不影响上式因此现在就是要利用变分法,看在限制i j ij f f δ=下,什么样i f 的会使E 最小,所以要利用Lagrange 乘子法:**()Nij i j ij ij iji ij ij Nij i jij ij iji j i j j i ij ij ji ij L E f f f L E f f L f f f f f f εεδεδεεεεεεε=--=-=∴=∑∑ 对变分,为常数,可不管。
分⼦轨道理论分⼦轨道理论同核双原⼦分⼦如您所知,电⼦在原⼦中存在于不同能级(例如1s,2s,3d等)的轨道中。
这些轨道表⽰在原⼦周围任何地⽅找到电⼦的概率分布。
分⼦轨道理论提出了这样⼀个概念,即分⼦中的电⼦同样存在于不同的轨道中,这使⼈们有可能在分⼦周围的特定点找到电⼦。
为了产⽣分⼦的轨道集,我们将分⼦中键合原⼦的价原⼦波函数加在⼀起。
这并不像听起来那样复杂。
让我们考虑同核双原⼦分⼦中分⼦式A 2的键合。
也许我们能想到的最简单的分⼦是氢H 2。
正如我们已经讨论过的,要产⽣氢每个氢的分⼦轨道,我们将价原⼦波函数加在⼀起以产⽣氢的分⼦轨道。
H 2中的原⼦仅具有1s轨道,因此我们将两个1s波函数相加。
正如您在原⼦结构研究中所了解的那样,原⼦波函数可以具有正或负相位-这意味着波函数y的值可以为正或为负。
有两种添加波函数的⽅法:同相(正负两个)或异相(正负另⼀个)。
展⽰了如何将原⼦波函数加在⼀起以产⽣分⼦轨道。
图%:两个1s轨道结合形成键和反键MO同相重叠组合(中的顶部轨道)在两个原⼦核之间产⽣电⼦密度的累积,从⽽导致该轨道的能量较低。
占据s H-H轨道的电⼦代表H 2的Lewis结构的电⼦键对,并适当地称为键分⼦轨道。
产⽣的另⼀个分⼦轨道s * HH显⽰原⼦核之间的电⼦密度降低,在存在节点平⾯的原⼦核之间的中点达到零值。
由于s * HH轨道显⽰出两个原⼦核之间键合的减少,这被称为反键分⼦轨道。
由于原⼦核之间电⼦密度的降低,抗键合轨道的能量⾼于键合轨道和氢1s轨道。
在分⼦H 2,没有电⼦占据反键轨道。
中总结这些关于键,反键和原⼦轨道的相对能量的发现,我们可以构建⼀个轨道相关图,如下所⽰:图%:氢的轨道相关图请注意,分离的原⼦的轨道写在图的两侧,是⽔平线,其⾼度表⽰它们的相对能量。
每个原⼦轨道上的电⼦⽤箭头表⽰。
在图的中间,写下了感兴趣分⼦的分⼦轨道。
虚线将母原⼦轨道与⼦分⼦轨道连接起来。
通常,键合分⼦轨道的能量低于其母原⼦轨道中的任何⼀个。
分子轨道理论及基态与激发态分子轨道理论基本概念一、分子轨道:(molecular orbital) 描述分子中电子运动的波函数,指具有特定能量的某电子在相互键合的两个或多个原子核附近空间出现的概率最大的区域。
分子轨道由原子轨道线性组合而成。
二、成键三原则:能量相近、最大重叠、对称性匹配。
只有对称性相同的两个原子轨道才能组成分子轨道。
6对称:一个原子轨道,取X轴作为对称轴,旋转180°,轨道符号不变。
如S,Px,d x2-y2 为6对称。
n对称:一个原子轨道,取X轴作为对称轴,旋转180°,轨道符号改变。
Py, Pz, d xy 是n对称。
由6对称的原子轨道组成的键——6键由n对称的原子轨道组成的键-- n键三、成键轨道与反键轨道分子轨道与原子轨道的联系:轨道守恒——2个原子轨道线性组合,产生 2 个分子轨道;能量守恒——2个分子轨道的总能量等于 2 个原子轨道的总能量;能量变化——每个分子轨道的能量不同于原子轨道的能量组合结果—定会出现能量高低不同的两个分子轨道。
——这是原子轨道线性组合的方式不同所致。
波函数同号的原子轨道相重叠,原子核间的电子云密度增大,形成的分子轨道的能量比各原子轨道能量都低,成为成键分子轨道。
波函数异号的原子轨道相重叠,原子核间的电子云密度减小,形成的分子轨道的能量比各原子轨道能量都高,成为反键分子轨道。
四、电子填入分子轨道时服从以下原则:1、能量最低原理:电子在原子或分子中将优先占据能量最低的轨道。
2、保利不相容原理:在同一原子或分子中、同一轨道上只能有两个电子,且自旋方向必须相反。
3、洪特规则:在能量相同的轨道中(简并轨道),电子将以自旋平行的方式、分占尽可能多的轨道基态与激发态当分子中的所有电子都遵从构造原理的这三个原则时,分子所处的最低能量状态——基态。
通常情况下,分子处于基态。
激发态:当分子获取能量后,分子中的电子排布不完全遵从构造原理,分子处于能量较高的状态——激发态,是原子或分子吸收一定的能量后,电子被激发到较高能级但尚未电离的状态。
分子轨道理论简介一种化学键理论,是原子轨道理论对分子的自然推广。
其基本观点是:物理上存在单个电子的自身行为,只受分子中的原子核和其他电子平均场的作用,以及泡利不相容原理的制约;数学上则企图将难解的多电子运动方程简化为单电子方程处理。
因此,分子轨道理论是一种以单电子近似为基础的化学键理论。
描写单电子行为的波函数称轨道(或轨函),所对应的单电子能量称能级。
对于任何分子,如果求得了它的系列分子轨道和能级,就可以像讨论原子结构那样讨论分子结构,并联系到分子性质的系统解释。
有时,即便根据用粗糙的计算方案所得到的部分近似分子轨道和能级,也能分析出很有用处的定性结果。
理论⒈原子在形成分子时,所有电子都有贡献,分子中的电子不再从属于某个原子,而是在整个分子空间范围内运动。
在分子中电子的空间运动状态可用相应的分子轨道波函数ψ(称为分子轨道)来描述。
分子轨道和原子轨道的主要区别在于:⑴在原子中,电子的运动只受1个原子核的作用,原子轨道是单核系统;而在分子中,电子则在所有原子核势场作用下运动,分子轨道是多核系统。
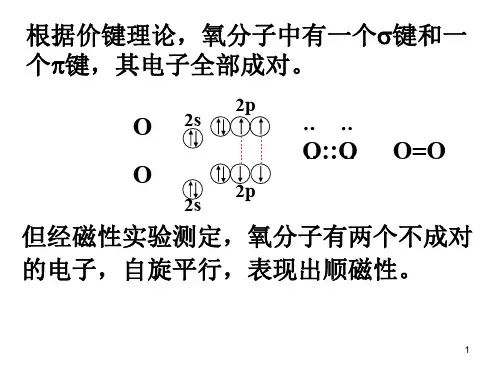
分子轨道理论⑵原子轨道的名称用s、p、d…符号表示,而分子轨道的名称则相应地用σ、π、δ…符号表示。
⒉分子轨道可以由分子中原子轨道波函数的线性组合(linearcombinationofatomicorbitals,LCAO)而得到。
有几个原子轨道就可以可组合成几个分子轨道,其中有一部分分子轨道分别由对称性匹配的两个原子轨道叠加而成,两核间电子的概率密度增大,其能量较原来的原子轨道能量低,有利于成键,称为成键分子轨道(bondingmolecularorbital),如σ、π轨道(轴对称轨道);同时这些对称性匹配的两个原子轨道也会相减形成另一种分子轨道,结果是两核间电子的概率密度很小,其能量较原来的原子轨道能量高,不利于成键,称为反键分子轨道(antibondingmolecularorbital),如σ*、π*轨道(镜面对称轨道,反键轨道的符号上常加"*"以与成键轨道区别)。
第五章分子轨道理论5.1 Hatree-Fock 方程Hatree-Fock 近似,也就是分子轨道近似,是量子化学中心之一,分子中的电子占据轨道,这是化学家头脑中很容易想到的。
首先,我们推导一下Hatree-Fock 方程。
由于绝大多数分子都是闭壳层的,因此我们都可以用单slater 行列式作为其波函数,即12N C f f f ψ=设我们有正交集i j ij f f δ= 则一、二阶约化密度矩阵为:'*'11111''111112''21212''112122(,)()()(,)(,)1(,;,)2(,)(,)i i ix x f x f x x x x x x x x x x x x x ρρρρρρ∧∧∧∧∧∧==∑改写一下(Dirac ):*'*'11122*'*'2122''1212()()()()12()()()()1[()()()()]2NNi i i i iiNNj j j j jjN i j i j i jj i i jf x f x f x f x f x f x f x f x f x f x f x f x f f f f ρ∧≠==-∑∑∑∑∑12(1)(1,2)1(1)[(1,2)(1,2)]2(1,2)(1,2)1[]2r r Ni i i j i j i j j i ii ji i i ii i i i Ni i i j i j i j j i iijE T h T g f h f f f g f f f f g f f f f g f f f f g f f E f h f f f g f f f f g f f ρρ∧∧∧∧≠=+=+--=+-∑∑∑∑因为i=j 时,=0不影响上式因此现在就是要利用变分法,看在限制i j ij f f δ=下,什么样i f 的会使E 最小,所以要利用Lagrange 乘子法:**()Nij i j ij ij iji ij ij Nij i jij ij iji j i j j i ij ij ji ij L E f f f L E f f L f f f f f f εεδεδεεεεεεε=--=-=∴=∑∑ 对变分,为常数,可不管。
其中为Lagrange 乘子直接作变分可以,也可以先将[]矩阵对角化:实的:厄米的,即[]矩阵为厄米的,即[]为厄米矩阵,总可以找到Unitary 酋矩阵U 使之对角化定义新基底{}'if ,它与{if 有酋交换关系:'''''1i i j ji j j ijj ijjf U f f f U f U f f f Uf U ++-=====∑∑∑及写成矩阵元形式:这样,1[]2i i i j i j i j j i iijij i jijL f h f f f g f f f f g f f f f ε=+--∑∑∑在新基底下:''1''''11''''111111[2]11[2j j ji ij k l k l ki lj ik jlijijklk l l k ki lj ik jl ij i j ki jlijkljiij ki ij jl ki ij jl k kli iiN Ni i i j i j i j j iij E f h f U U f f g f f U U U U f f g f f U U U U f f U U UU U U U U L L f h f f f g f f f f g f f εεεεδ--------+=+--====+-∑∑∑∑∑∑∑∑且则去掉'号有:]Ni i i iijf f ε-∑现在进行变分:0L δ=0[()]i i j i j j j i i ii jiL f h f f g f f f g f f fδδε==+-++=∑∑∑共轭项式子中全体,共轭项为0又因电子相互独立,坐标不相关,因此求和中每一项须为0,即()i j i j j j i i ijh f f g f f f g f f fε+-=∑此式即为Hatree-Fock方程。
形式的写出来:()()Fi i iFh i f fh iε=其中为Hatree-Fock算子若定义算子iJ(库伦算子)及iK(交换算子)*12212*12212*212212()()(1,2)()()()()(1,2)()()()(1,2)[()()](1)(1)(1)-(1)Ni i j j ijNi i j i jjNj j ijNi i j j ijNi i j i jjFi iFiJ f x f x g f x f x dxK f x f x g f x f x dxf xg P f x f x dxJ f f g f fK f f g f fHFh h J Kh f======+∑⎰∑⎰∑⎰∑∑即则方程有形式:i iij iifHatree Fockfεεε=-称为正则方程(因[]对角化至得来)为正则分子轨道。
5.2 Hatree-Fock方程解的性质①(1)Fh为厄米算子{}{}*22122*22122(1)(1)()(1)()(1)(1)(()(1)())(1)(1)-F j j jF j j jF F F i i i iF i i i i h h f x gdx P f x h h f x gdx f x h h h f f f h f f ρεεε++++=+-=+-∴==∴=∑⎰∑⎰F 则为厄米的,应有本征函数完全集Hatree-Fock 方程,构成本征函数完全集,相应的本征值为,可看出:是由轨道决定的,h 为一个积分微分方程,对于分子体系一般是不可解的,以后我们要讨论求解HF 方程(Roothaan 的工作)②HF 算子的轨道及轨道能量 HF 算子的本征函数i f 为轨道对于一个N 电子体系,将i ε排序12εε<那么HF 算子的前N 个轨道为占据轨道,一般用,,a b c 大于N 的轨道为非占据轨道,或虚轨道,,,r s i ε称为轨道能量。
轨道总能量:[]F i i iiNi i i j i j i j j i i ijf h ff h f f fg f f f f g f f εε===+-∑∑∑∑而刚才我们已经给出了体系的能量(用密度矩阵方法)101[]2,[][][]Ni i i j i j i j j i iijNa b Na b a Nr b aE f h f f f g f f f f g f f E E a h a ab g ab ab g ba a h a ab g ab ab g ba N r h r rb g rb rb g br εεεεψε=≠≠=+-≠>=+-∴=+-⎫⎪⎪⎪⇒⎬⎪⎪⎪⎭=+-∑∑∑∑∑ 占据轨道能量:当a=b 时,上式第二项为——个占据轨道基态,单slater 行列式————b ——a 虚轨道能量:③Koopmans ’定理 概念:冻结轨道近似:加入或拿掉一个轨道上的电子,并不影响其它轨道,N 电子体系总能量为: 1[]2NNaabE a h a ab g ab ab g ba =+-∑∑,N-1电子体系总能量为:11[]2NN a ca cb cE a h a ab g ab ab g ba -≠≠≠=+-∑∑即去掉c 轨道则体系电离势为1N N c IP E E ε-=-=-证明:NE 重写N-1为部分 + 一个带(c )的部分:1111[]21[]21[]2()[]1[2NNa ca cb cNa cb cNa cb cNN Nca cN N rN aE a h a c h c ab g ab ab g ba g cb cb g bc ac g ac ac g ca Ionization Potential IP IP E E c h c ac g ac ac g ca E E E a h a r h r ab εε≠≠≠=≠≠=-≠++=++-+-+-=-=---=--=-=++∑∑∑∑∑∑从而 为:并且,体系电子亲和势可类似求出:即11]1[]21[]2[]000,0Nab NaNbNN N rac r N N r g ab ab g ba ar g ar ar g ra rb g rb rb g br E E r h r ar g ar ar g ra E E εεεε++-+-+-∴-=---=-<∴><>>∑∑∑∑一般电离能而为虚轨道,不好说。
若则电子亲和势,即,则N+1体系比N 体系更稳定。
上面两条式子就是Koopmans 定理的内容。
它将电离势与电子亲和势与轨道能量联系起来,可是实际应用中有局限性。
Koopmans 定理局限性:1. 忽略了电子重排——松弛效应2. 忽略了电子相关3. 忽略了相对论效应所以,电离势的计算,若要尽量准确的话,还需要加上对2,3的修正。
但在Hatree-Fock 方程基础上,我们只能最多对1进行修正。
文献中有两种电离势定义: 垂直电离势:即离子与分子都用分子的几何(来修正)绝热电离势:,M M +分别优化(修正,考虑松弛) ④Brillouin 定理 薛氏方程H E ψψψ∧=为H 真实,准确基态而HF 方法用单slater 行列式0ψ来近似ψ,H ∧在0ψ下有平均值000H E ψψ∧=00000001000,():NF i aaHatree Fock Hamiltonian H H HF Hamiltonian H h i H ψψεψεεε∧∧∧=∧-===∑∑其实为的本征函数其中:本征值,我们要求体系真实基态,必须包括其它形式的slater 行列式,即对HF 基态修正,我们总可以写成:00,0000000001[]rr a a r ar a r a r ra a r a r r r a aara Nb Fa a c c c c H H H H H a h r ab g rb ab g br a h r a r ψψψψψψψψψψψψψψψψψψψψεεδ∧∧∧∧∧==++=+⎛⎫ ⎪ ⎪ ⎪⎝⎭=+-===∑∑其中为HF 基态为中a 被虚轨道r 取代,为第一激发态,现在我们只考虑某一单激发态,则其对应的本征方程为HC=EC 我们现在来求Hamiltonian 矩阵:H=00000arrr aa H H H ψψψψ∧∧⎛⎫ ⎪ ⎪ ⎪⎝⎭所以矩阵中,非对角元为,则H 为:H=也即HF 基态在某种意义上“稳定的”,因其不能由考虑单激发态而得到修正。