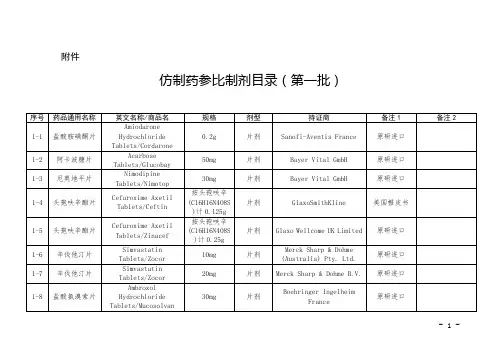
化学仿制药参比制剂目录(第三十二批)(征求意见稿)
- 格式:docx
- 大小:53.63 KB
- 文档页数:12

国家食品药品监督管理总局关于征求化学仿制药生物等效性试验备案管理规定(征求意见稿)意见的公告文章属性•【公布机关】国家食品药品监督管理总局•【公布日期】2015.11.06•【分类】征求意见稿正文国家食品药品监督管理总局关于征求化学仿制药生物等效性试验备案管理规定(征求意见稿)意见的公告(国家食品药品监督管理总局公告2015年第221号)为贯彻落实《国务院关于改革药品医疗器审评审批制度的意见》(国发〔2015〕44号),简化药品审批程序,食品药品监管总局组织起草了《化学仿制药生物等效性试验备案管理规定(征求意见稿)》,现向社会公开征求意见。
请将修改意见于2015年11月20日前通过电子邮件反馈至食品药品监管总局。
联系人:王晓刚、柳雨时联系电话:************电子邮箱:**************.cn特此公告。
附件:化学仿制药生物等效性试验备案管理规定(征求意见稿)国家食品药品监管总局2015年11月6日附件化学仿制药生物等效性试验备案管理规定(征求意见稿)第一章总则第一条为落实药品审评审批制度改革要求,优化仿制药审批流程,根据《中华人民共和国药品管理法》《中华人民共和国药品管理法实施条例》《药品注册管理办法》《药物非临床研究质量管理规范》《药物临床试验质量管理规范》等,制定本规定。
第二条在中华人民共和国境内以药品注册、仿制药质量疗效一致性评价为目的开展化学仿制药生物等效性试验(BE试验)的备案和监督管理,适用本规定。
第三条化学仿制药BE试验备案,是指化学仿制药BE试验备案申请人(以下简称申请人)在化学仿制药研究过程中,在国家食品药品监督管理总局指定的信息平台按照要求提交备案资料,获得备案号后自行开展并完成BE试验的全过程。
第四条申请人提出备案申请并开展BE试验属于药品申请受理前的自主研制阶段,申请人应按照药品注册或仿制药质量疗效一致性评价的相关法律法规和技术要求开展研究,对研究的合法性、合理性、科学性、伦理符合性承担全部责任。
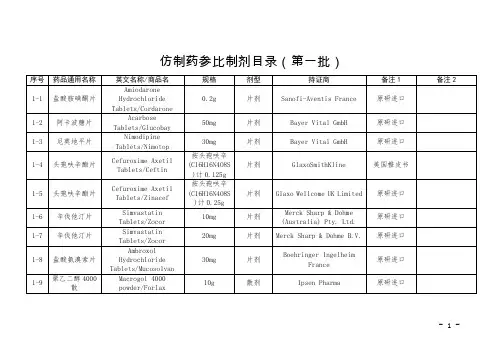
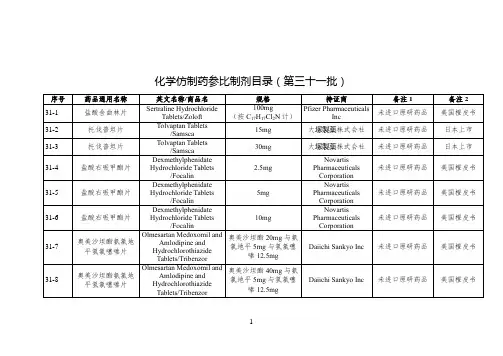
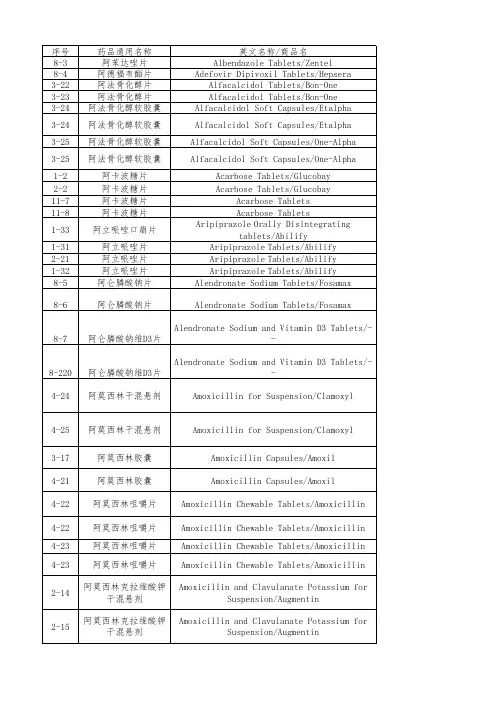

国家药监局关于发布仿制药参比制剂目录(第三十
批)的通告
文章属性
•【制定机关】国家药品监督管理局
•【公布日期】2020.08.31
•【文号】国家药品监督管理局通告2020年第56号
•【施行日期】2020.08.31
•【效力等级】部门规范性文件
•【时效性】现行有效
•【主题分类】药政管理
正文
国家药品监督管理局通告
2020年第56号
国家药监局关于发布仿制药参比制剂目录(第三十批)的通
告
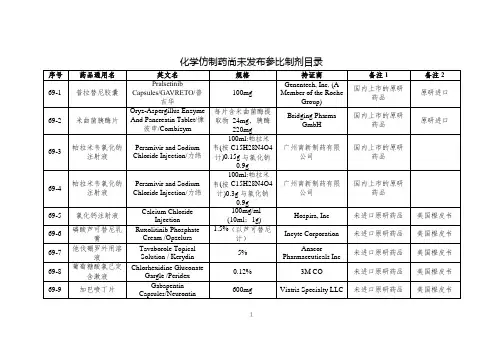
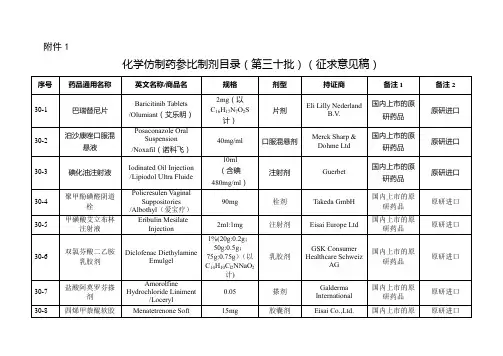
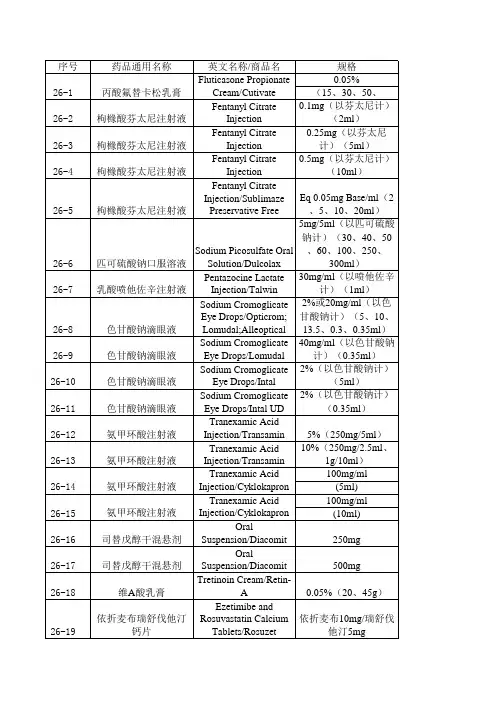
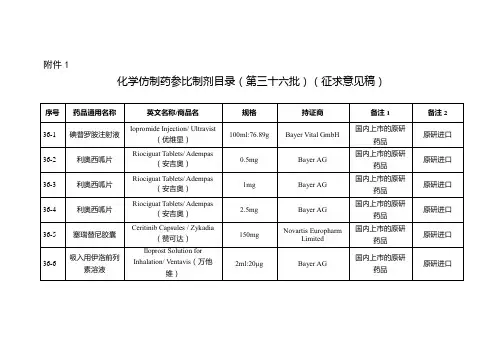
经国家药品监督管理局仿制药质量和疗效一致性评价专家委员会审核确定,现发布仿制药参比制剂目录(第三十批)。
特此通告。
附件:仿制药参比制剂目录(第三十批)
国家药监局
2020年8月31日。

附件仿制药质量与疗效一致性评价参比制剂备案与推荐程序(征求意见稿)为贯彻落实国务院办公厅《关于开展仿制药质量和疗效一致性评价的意见》(国办发〔2016〕8号)的任务要求,进一步明确参比制剂的选择流程,制定本备案与推荐程序。
一、本程序所述参比制剂备案与推荐工作,是指药品生产企业、行业协会、原研药品生产企业、国际公认的同种药物生产企业等作为申请人或推荐人,参照《普通口服固体制剂参比制剂选择和确定指导原则》,通过备案、推荐、申报等方式,选择参比制剂的过程。
二、药品生产企业可通过备案的方式选择参比制剂。
生产企业填写《参比制剂备案表》(附件1);撰写《综述资料》(附件4),详述参比制剂选择理由;提交所生产品种现行有效的批准证明文件,生产产品首次批准和上市后变更等历史沿革情况的说明。
三、行业协会可组织同品种生产企业提出参比制剂的推荐意见。
行业协会填写《参比制剂推荐表》(附件2);撰写《综述资料》(附件4),详述参比制剂选择理由;提交行业协会资质证明复印件、推荐过程记录与说明、相关同品种生产企业同意推荐的证明性文件。
四、原研药品生产企业、国际公认的同种药物生产企业其产品如可满足参比制剂的条件,可主动申报作为参比制剂。
生产企业填写《参比制剂申报表》(附件3);撰写《综述资料》(附件4),详述参比制剂选择理由;提供申报参比制剂品种近三年生产、销售情况说明。
进口原研药品申报参比制剂,申报者需同时提交生产证明、销售证明、出口证明等证明文件,及进口原研药品与其原产国上市药品一致的承诺书。
原研地产化药品申报参比制剂,申报者需同时提交原研地产化药品与原研药品一致的相关证明材料,具体要求见《原研地产化产品申报口服固体制剂参比制剂资料要求》(附件5)。
五、申请人或推荐人应对提交资料的真实性负责。
纸质版邮寄至中国食品药品检定研究院仿制药质量研究中心,并标注“一致性评价参比制剂备案与推荐材料”;电子版同时发送至cbzjbatj@,邮件标题为“申请人名称—参比制剂备案与推荐”。
化学仿制药参比制剂遴选申请资料要求(征求意见稿)一、申请综述(一)品种基本信息1.药品通用名称2.药物活性成分名称3.剂型和规格4.适应症简述5.各国药典收载情况简要说明各国药典收载情况,并注明药典出版时间或版本号。
6.参比制剂发布/公示情况国家药品监督管理局已于第xx批《化学仿制药参比制剂目录》中发布本品种参比制剂,详见下表。
或本品国家药品监督管理局尚未发布参比制剂。
表xx:本品种已发布/已公示参比制剂情况表7.1原研是否明确是/否。
简要概述原研产品上市情况(建议包括但不限于原研企业,上市时间、最早上市国家、商品名、剂型、规格等)。
如持证商发生变更,请说明时间节点和变更情况。
应提供原研调研信息及相关证明性文件。
7.2临床安全有效性是否明确是/否。
简要概述原研药品上市前、后临床研究信息。
综上,本品属于(原研明确、疗效明确;原研不明、疗效明确;原研明确、疗效不明;原研不明、疗效不明)类化学药品。
(二)参比制剂申请依据根据《化学仿制药参比制剂遴选与确定程序》进行综合评估,包括但不限于以下内容:1.参比制剂申请原因如国家药品监督管理局尚未发布参比制剂,或对已发布参比制剂存在异议(需明确具体原因,并提供相关证明性文件)。
2.拟申请参比制剂是否为原研应明确拟申请参比制剂是否为原研,持证商,以及来源。
3.拟申请参比制剂上市时是否有完整和充分的安全、有效性数据如拟申请参比制剂为原研产品,则此项内容可填写为同“(一)品种基本信息下7.2项”,无需赘述。
如拟申请参比制剂为其他国际公认产品,需简要评价产品作为参比制剂的依据及其安全有效性。
4.拟申请参比制剂的质量是否可控、是否符合现行的国际通用技术要求(如ICH等)如尚未开展相关研究,说明情况即可。
如已有研究数据,可简要概述拟申请参比制剂质量是否可控。
5.拟申请参比制剂是否可及提供销售数据或其他能够证明可及性的文件均可。
6.拟申请参比制剂在美、欧、日及国内上市的处方是否一致结合调研信息,简要概述拟申请参比制剂在美、欧、日及国内的处方情况,明确是否一致,如不一致可简要分析处方差异对产品的影响。