陕西师范大学物理化学精品课程
- 格式:pdf
- 大小:168.76 KB
- 文档页数:7

复习试题一1.有一系统在某一过程中,吸热30J ,对外做功50J ,则内能变化量为 ;2.已知反应C(s)+O 2(g)=CO 2(g) 的标准平衡常数为K 1;CO(g)+1/2O 2(g)=CO 2(g)的标准平衡常数为K 2; 2C(s)+O 2(g)=2CO(g) 的标准平衡常数为K 3;则K 3与K 1、K 2的关系为 _____________ ; 3.某理想气体在20℃、100kPa 下的摩尔体积为 ;4.焓、吉布斯自由能的定义式分别为 、;1.下列各种条件下,CO 2在水中溶解度最大的是( )A 高压低温B 低压低温C 高压高温D 低压高温2.在 400K ,液体A 的蒸气压为4×104 Pa ,液体B 的蒸气压为6×104 Pa ,两者组成理想液态混合物。
当气-液平衡时,在溶液中A 的摩尔分数为0.6,则在气相中B 的摩尔分数应为( ) A 0.31 B 0.40 C 0.50 D 0.603.在0.1 kg 水中含 0.0045 kg 某纯非电解质的溶液,于272.685 K 时结冰,水的凝固点降低常数 K f 为1.86 K.mol -1.kg -1,则该溶质的摩尔质量大约为( ) A 0.135 kg/mol B 0.172 kg/molC 0.090 kg/molD 0.180 kg/mol4. PCl 5的分解反应是PCl 5(g )=PCl 3(g) + Cl 2(g),在473 K 达到平衡时, PCl 5(g)有48.5%分解,在573 K 达到平衡时,有97%分解,则此反应为( )A 放热反应B 吸热反应C 即不放热也不吸热D 这两个温度下的平衡常数相等一、填空题(共4小题,将适当的内容填入题中划线处。
每空3分,满分15分)二、选择题( 共4题,将判断结果填入题后的括号中。
每题3分,满分12分)( )1. 某一化学反应的标准摩尔吉布斯函数△r G m $> 0,这就说明该反应不能自发进行; ( )2. 在通常情况下,对于二组分系统平衡共存时最多相数为3; ( )3. 米和面混合的十分均匀,再也无法彼此分开,则该系统有一个相; ( )4. 水的三相点和水的冰点不是一回事;( )5. 标准平衡常数改变了,平衡一定会移动。




物理化学实验课程中学生综合能力的培养薛敏;杨菊香;刘振【摘要】物理化学实验课是化学相关专业的一门重要的基础课程.探讨了如何在物理化学实验教学中培养学生的综合能力.结合具体的教学过程,重点从培养学生的动手能力、数据处理能力以及创新能力等几个方面介绍了一些方法和经验.实践证明,采用这些方法实施教学,大大的提高了学生的学习兴趣,培养了学生各方面的综合能力.【期刊名称】《广州化工》【年(卷),期】2013(041)012【总页数】2页(P222-223)【关键词】物理化学实验;综合能力;创新思维【作者】薛敏;杨菊香;刘振【作者单位】西安文理学院化学与化学工程学院,陕西西安710065;西安文理学院化学与化学工程学院,陕西西安710065;西安文理学院化学与化学工程学院,陕西西安710065【正文语种】中文【中图分类】G642.423物理化学实验是化学化工及其相关专业的一门理论性和实践性都很强的基础课程,是化学实验学科的重要分支,它综合了化学领域中各分支学科所需要的基本研究技术和方法。
学生在上完无机化学实验、分析化学实验和有机化学实验课程后的最后一门基础实验课就是物理化学实验。
物理化学实验课程的主要目的是巩固并加深对物理化学理论课所学知识的理解;掌握物理化学实验的基本方法和物理化学实验仪器的使用;培养学生动手能力、思维能力和分析实验结果的能力等。
但是,如何才能在物理化学实验课程的教学中,真正实现以上的目的,并且能够培养出高素质、创新性、能够适应现代社会激烈竞争的综合性人才,是所有教师都面临的重要课题[1-3]。
为了锻炼学生的各种综合能力,为了培养出高素质的综合性人才,使学生在走上工作岗位或继续深造时能够具有更强的竞争力,我们在物理化学实验教学中做出了一定的尝试,具体阐述如下。
1 注重学生动手能力的培养物理化学实验课要求学生在深入理解物理化学相关理论知识的基础上,还必须具有很强的动手能力和使用仪器的能力。
通过物理化学实验课程,培养学生的动手能力,了解各种仪器的构造、功能和使用,使学生在毕业后能更顺利的完成工作或科研任务,在激烈的竞争中立于不败之地。

建设高水平教学团队不断提升课程教学质量摘要:陕西师范大学物理化学教学团队针对传统物理化学课程在创新性人才培养需求的局限性,构建以物理化学基础课程群和拓展课程群为特色的现代物理化学教学体系,提高学生对物理化学及相关课程的关联性认识和物理化学理论与其实际应用相结合的能力;以物理化学为核心整合相关课程师资队伍,强化教学团队整体实力;以改进传统物理化学实验与创建学生深入教师科研的创新性实验教学模式相结合,提升学生综合素养;以扩大学术交流,提升教学团队水平和学术影响力。
上述教学团队建设实践对于教学改革具有积极的借鉴意义。
关键词:物理化学;教学团队;教学改革2007年初,教育部提出“加强本科教学团队建设,重点遴选和建设一批教学质量高、结构合理的教学团队,建立有效的团队合作机制,推动教学内容与教学方法的改革和研究,促进教学研讨和教学经验交流,开发教学资源,推进教学工作的老中青相结合,发扬传、帮、带的作用,加强青年教师培养”。
这一举措对高校教学质量提高起到了特别积极的作用。
陕西师范大学物理化学(含结构化学)教学团队坚持“以学科核心凝聚团队力量,以人才培养新需求开拓创新”的团队建设与发展理念,经过多年努力,教学团队建设取得了显著成绩。
该教学团队开设的物理化学课程先后被评为陕西省精品课程、国家精品课程。
2008年物理化学(含结构化学)教学团队获国家级教学团队称号。
2009年结构化学被评为省级精品课程。
教学团队建设一方面促进了教学水平的提高,另一方面也使教学团队的学术影响力得到显著提升。
一、适应创新人才培养需求,构建新的教学体系学生的创新能力培养得到社会广泛关注,然而,如何在教学中实施创新能力的培养仍然是当今高等教育的主题嘲。
我们认为,创新人才就是具有创新素质的人。
创新素质是在人的基本素质基础上形成的一种能够用灵活多样的方式去创造新事物、解决新问题的高级的、复杂的、综合的能力素质。
创新人才应达到以下基本要求:(1)扎实的专业基础知识和灵活运用专业知识的能力。

第七章电解质溶液本章内容:介绍电化学的基本概念和法拉第定律、离子的电迁移数、测定迁移数的常用方法;电导、电导率、摩尔电导率的意义;离子独立移动定律及电导测定的一些应用,强电解质溶液理论(主要是离子氛的概念),并会使用德拜-休克尔极限公式。
第一节电化学的基本概念和法拉第定律一、基本概念1. 导体:能导电的物质称为导电体简称导体(1) 第一类导体: 电子导体(如金属、石墨及某些金属的化合物等)。
导电机理: 靠自由电子的定向运动而导电,在导电过程中本身可能发热,但不发生化学变化。
特性: 随温度的升高,由于质点的热运动加剧,阻碍了自由电子的定向运动,因而电阻增大,导电能力降低。
(2) 第二类导体: 离子导体(如电解质溶液或熔融的电解质等)。
导电机理: 靠离子的定向运动而导电,即依赖正、负两种离子各向反方向迁移以运输电量,当插入电解质溶液中的两电极间存在电位差时,正离子移向阴极,负离子移向阳极,同时在电极上有化学变化发生。
特性: 温度升高时,由于溶液的粘度降低,离子运动速度加快,在水溶液中离子水化作用减弱等原因,导电能力增强。
2. 电池: 由第一类导体联结两个电极并使电流在两极间通过,构成外电路的装置叫做电池。
3.电解池: 在外电路中并联一个有一定电压的外加电源,则将有电流从外加电源流入电池,迫使电池中发生化学变化,这种将电能转变为化学能的电池称为电解池4.原电池: 电池能自发地在两极上发生化学反应,并产生电流,此时化学能转化为电能,则该电池就称为原电池。
5.正极和负极: 电势较高的极称为正极,电势较低的极称为负极。
电流总是由正极流向负极,电子的流向与之相反。
6.阳极和阴极: 发生氧化反应的电极称为阳极,发生还原反应的电极称为阴极。
两种电化学装置的正、负极和阴、阳极之间的对应关系: 在电解池中,与外电源负极相接的电极接受电子,电势较低,发生还原反应,所以该电极是负极也是阴极;与外加电源正极相接的电极,电势较高,发生氧化反应,所以该电极是正极也是阳极。



陕西师范大学物理化学精品课程第十二章 界面现象本章内容:介绍表面吉布斯自由能和表面张力的概念;固体表面吸附的兰缪尔公式和BET 公式;明确弯曲表面附加压力产生的原因,介绍杨-拉普拉斯公式和开尔文公式;了解一些常见的表面现象和表面活性剂的作用。
第一节 表面吉布斯自由能和表面张力一、表面吉布斯自由能和表面张力由于表面层分子的状咬与本体中不同,因此如果要把一个分从内部移到界面(或者说增大表面积)时,就必须克服体系内部分子这间的吸引力而对体系作功。
在温度、压力和组成恒下时,可逆地使表面积增加d A 所需要对体系做的功,叫做表面功,可以表示为A W d f γ=δ ( 12–1 )式中是比例常数,它在数值上等地当T、p 及组成恒定的条件下,增加单位表面积时所必体系做的可逆非膨胀功。
γ由于,所以上式又可以表示为p T,G W d f =δ A G p T,d d γ= 或 p T,AG )(∂∂=γ ( 12–2 ) 可以看出,的物理意义是:在定温不定定压条件下,可逆地增加单位表面积引起体系吉布斯自由能的增量。
单位为。
γ2m J −⋅γ称为表面吉布斯自由能或比表面能。
从另一个角度来考虑,如果观察到表面上处处存在着一种张力,称之为表面张力(surface tension )。
它作用在表面的边界线上,垂直于边界线向着表面的中心并与表面相切,或者是作用在液体表面上任一条线的两侧,垂直于该线,沿着液面拉向两侧。
例如,把金属丝弯成U 形框架,放在肥皂膜,由于表面张力的作用,会肥可滑动的金属丝拉上去,一直到框架顶部,如在金属丝下面吊一重物W ,如果与可滑动金属丝的质量W 之和(即W +W)与向上的表面张力平衡时,金属丝就保持不同志滑动。
22W 112在图12.2中,虽然肥皂膜很薄,但和分子的大小相比,还具有一定的厚度,可以认为肥皂膜有一定的体积,在金属丝框架的正反两面具有两个表面,所以表面张力在总长度为的边界上作用,由于表面上张力的指垂直地作用于单位长度的表面边沿,并指向表面中心的力,所以肥皂膜将金属丝向上拉的力(即等于向下的重力(+)g)为2γ1W 2W 12= 2= ( + ) F l W W g γ这里称为表(界)面张力,其单位为,这是从另一角度来理解的(表面自由能的单位是,由于,所以的单位也可表示为,N 为牛顿,是力的单位,所以表面自由能也可以看作是垂直用于单位长度相界面上的力即表面张力)。
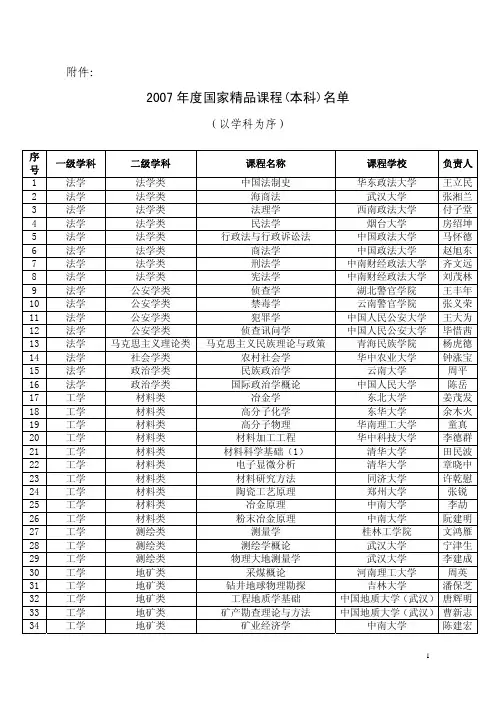
附件:2007年度国家精品课程(本科)名单(以学科为序)序一级学科 二级学科 课程名称 课程学校 负责人号1 法学 法学类 中国法制史 华东政法大学 王立民2 法学 法学类 海商法 武汉大学 张湘兰3 法学 法学类 法理学 西南政法大学 付子堂4 法学 法学类 民法学 烟台大学 房绍坤5 法学 法学类 行政法与行政诉讼法 中国政法大学 马怀德6 法学 法学类 商法学 中国政法大学 赵旭东7 法学 法学类 刑法学 中南财经政法大学 齐文远8 法学 法学类 宪法学 中南财经政法大学 刘茂林9 法学 公安学类 侦查学 湖北警官学院 王丰年10 法学 公安学类 禁毒学 云南警官学院 张义荣11 法学 公安学类 犯罪学 中国人民公安大学 王大为12 法学 公安学类 侦查讯问学 中国人民公安大学 毕惜茜13 法学 马克思主义理论类 马克思主义民族理论与政策 青海民族学院 杨虎德14 法学 社会学类 农村社会学 华中农业大学 钟涨宝15 法学 政治学类 民族政治学 云南大学 周平16 法学 政治学类 国际政治学概论 中国人民大学 陈岳17 工学 材料类 冶金学 东北大学 姜茂发18 工学 材料类 高分子化学 东华大学 余木火19 工学 材料类 高分子物理 华南理工大学 童真20 工学 材料类 材料加工工程 华中科技大学 李德群21 工学 材料类 材料科学基础(1) 清华大学 田民波22 工学 材料类 电子显微分析 清华大学 章晓中23 工学 材料类 材料研究方法 同济大学 许乾慰24 工学 材料类 陶瓷工艺原理 郑州大学 张锐25 工学 材料类 冶金原理 中南大学 李劼26 工学 材料类 粉末冶金原理 中南大学 阮建明27 工学 测绘类 测量学 桂林工学院 文鸿雁28 工学 测绘类 测绘学概论 武汉大学 宁津生29 工学 测绘类 物理大地测量学 武汉大学 李建成30 工学 地矿类 采煤概论 河南理工大学 周英31 工学 地矿类 钻井地球物理勘探 吉林大学 潘保芝32 工学 地矿类 工程地质学基础 中国地质大学(武汉)唐辉明33 工学 地矿类 矿产勘查理论与方法 中国地质大学(武汉)曹新志34 工学 地矿类 矿业经济学 中南大学 陈建宏号35 工学 电气信息类 编译原理 北京工业大学 蒋宗礼36 工学 电气信息类 数字图像处理 北京交通大学 阮秋琦37 工学 电气信息类 现代密码学 北京邮电大学 杨义先38 工学 电气信息类 嵌入式系统及应用 电子科技大学 罗蕾39 工学 电气信息类 现代电子技术实验 电子科技大学 习友宝40 工学 电气信息类 电力系统分析 东北电力大学 穆钢41 工学 电气信息类 电机学 东南大学 胡虔生42 工学 电气信息类 算法与数据结构 福州大学 王晓东43 工学 电气信息类 编译原理 国防科学技术大学 王挺44 工学 电气信息类 计算机网络 国防科学技术大学 徐明45 工学 电气信息类 光纤理论与技术 哈尔滨工程大学 苑立波46 工学 电气信息类 C语言程序设计 哈尔滨工业大学 王宇颖47 工学 电气信息类 集合论与图论 哈尔滨工业大学 王义和48 工学 电气信息类 电机学 哈尔滨理工大学 戈宝军49 工学 电气信息类 计算机组成原理 杭州电子科技大学 包健50 工学 电气信息类 电气工程实践 黑龙江科技学院 付家才51 工学 电气信息类 操作系统原理 华中科技大学 庞丽萍52 工学 电气信息类 电力电子学 华中科技大学 康勇53 工学 电气信息类 现代医学成像技术 南方医科大学 陈武凡54 工学 电气信息类 计算机通信与网络 南京邮电大学 杨庚55 工学 电气信息类 电工电子技术 青岛大学 徐淑华56 工学 电气信息类 电子商务概论 清华大学 覃征57 工学 电气信息类 软件工程 清华大学 孙家广58 工学 电气信息类 模式识别基础 清华大学 张学工59 工学 电气信息类 电路原理 清华大学 陆文娟60 工学 电气信息类 高电压工程 清华大学 梁曦东61 工学 电气信息类 自动控制理论 清华大学 王诗宓62 工学 电气信息类 基本电路理论 上海交通大学 陈洪亮63 工学 电气信息类 过程控制与集散系统 武汉科技大学 方康玲64 工学 电气信息类 微处理器类新技术实验 西安电子科技大学 石光明65 工学 电气信息类 软件开发技术基础 西安交通大学 顾刚66 工学 电气信息类 电工学 西北工业大学 史仪凯67 工学 电气信息类 自动控制原理 西北工业大学 卢京潮68 工学 电气信息类 电力牵引控制系统 西南交通大学 冯晓云69 工学 电气信息类 现代电子系统设计 西南科技大学 马建国70 工学 电气信息类 嵌入式系统 浙江大学 陈天洲71 工学 电气信息类 电力电子技术 浙江大学 潘再平72 工学 电气信息类 应用光学 浙江大学 岑兆丰73 工学 电气信息类 电子技术基础 中北大学 韩焱74 工学 工程力学类 理论力学 河海大学 武清玺75 工学 工程力学类 疲劳与断裂 华中科技大学 杨新华号76 工学 工程力学类 理论力学 昆明理工大学 屈本宁77 工学 工程力学类 流体力学 清华大学 符松78 工学 工程力学类 工程振动与测试 天津大学 刘习军79 工学 公安技术类 灭火战术 中国人民武装警察部队学院康青春80 工学 公安技术类 印刷文件检验 中国刑事警察学院 王世全81 工学 海洋工程类 船舶原理 上海交通大学 邹早建82 工学 航空航天类 导弹飞行力学 西北工业大学 方群83 工学 化工与制药类 化工原理 南京工业大学 居沈贵84 工学 化工与制药类 化工热力学 清华大学 高光华85 工学 化工与制药类 精细有机合成化学及工艺学 天津大学 冯亚青86 工学 化工与制药类 化学工艺学 天津大学 米镇涛87 工学 化工与制药类 制药工艺设计 武汉工程大学 张珩88 工学 环境与安全类 环境保护概论 北京工业大学 王淑莹89 工学 环境与安全类 水污染控制工程 桂林工学院 张学洪90 工学 环境与安全类 污染控制微生物学 哈尔滨工业大学 任南琪91 工学 环境与安全类 水污染控制工程 同济大学 周琪92 工学 机械类 画法几何及工程制图 大连理工大学 王殿龙93 工学 机械类 化工设备机械基础 大连理工大学 李志义94 工学 机械类 机械设计 东北大学 巩云鹏95 工学 机械类 工程制图 广东工业大学 左宗义96 工学 机械类 工程制图基础 国防科学技术大学 陈循97 工学 机械类 工程训练课程 哈尔滨工程大学 任正义98 工学 机械类 金属工艺学 哈尔滨工业大学 邢忠文99 工学 机械类 压力容器设计 华东理工大学 潘家祯100 工学 机械类 机械设计基础 华南理工大学 朱文坚101 工学 机械类 农业机械与设施 南京农业大学 丁为民102 工学 机械类 工程机械 石家庄铁道学院 杜彦良103 工学 机械类 机械制造技术基础 太原理工大学 吕明 104 工学 机械类 机械原理 武汉科技大学 孔建益105 工学 机械类 机械原理 西南交通大学 谢进 106 工学 机械类 工程制图 中国矿业大学 江晓红107 工学 交通运输类 城市轨道交通规划与设计 北京交通大学 毛保华108 工学 交通运输类 国际航运管理 上海海事大学 赵刚 109 工学 交通运输类 船舶辅机 武汉理工大学 向阳 110 工学 交通运输类 行车组织 西南交通大学 彭其渊111 工学 林业工程类 木材学 广西大学 罗建举112 工学 能源动力类 内燃机结构 哈尔滨工程大学 张文平113 工学 能源动力类 动力工程 华北电力大学 付忠广114 工学 能源动力类 传热学 清华大学 姜培学115 工学 能源动力类 工程流体力学 山东大学 杜广生号116 工学 能源动力类 传热学 上海理工大学 杨茉 117 工学 农业工程类 生物物料学 浙江大学 王俊 118 工学 轻工纺织食品类 纺纱学 东华大学 郁崇文119 工学 轻工纺织食品类 机织原理 江南大学 高卫东120 工学 轻工纺织食品类 制革整饰材料化学 陕西科技大学 马建中121 工学 轻工纺织食品类 食品工程原理 上海交通大学 李云飞122 工学 轻工纺织食品类 包装材料学 天津科技大学 王建清123 工学 轻工纺织食品类 葡萄酒工艺学 西北农林科技大学 李华 124 工学 轻工纺织食品类 成衣工艺学 浙江理工大学 邹奉元125 工学 轻工纺织食品类 食品保藏原理与技术 中国海洋大学 曾名湧126 工学 生物工程类 生物工艺学 北京化工大学 谭天伟127 工学 生物工程类 微生物学 浙江工业大学 沈寅初128 工学 水利类 水文统计 河海大学 黄振平129 工学 水利类 海岸动力学 河海大学 严以新130 工学 水利类 水工建筑学 清华大学 金峰 131 工学 土建类 结构设计原理 长沙理工大学 张建仁132 工学 土建类 建筑设计基础 华南理工大学 何镜堂133 工学 土建类 水力学 济南大学 于衍真134 工学 土建类 钢结构设计 青岛理工大学 王燕 135 工学 土建类 房屋建筑学 山东建筑大学 崔艳秋136 工学 土建类 中国古建筑测绘 天津大学 王其亨137 工学 土建类 建筑评论 同济大学 郑时龄138 工学 土建类 钢结构 西安建筑科技大学 郝际平139 工学 土建类 地下铁道 西南交通大学 高波 140 工学 土建类 建筑构造 重庆大学 魏宏杨141 工学 武器类 弹药学 南京理工大学 李向东142 工学 仪器仪表类 应用光学 长春理工大学 王文生143 工学 仪器仪表类 自动测试系统 电子科技大学 童玲 144 工学 仪器仪表类 测试与检测技术基础 清华大学 王伯雄145 工学 仪器仪表类 精密机械设计基础 天津大学 裘祖荣146 工学 仪器仪表类 传感器技术 中国计量学院 李东升147 管理学 工商管理类 中级财务会计 北京工商大学 杨有红148 管理学 工商管理类 物流系统论 北京工商大学 何明珂149 管理学 工商管理类 组织行为学 电子科技大学 井润田150 管理学 工商管理类 管理会计 东北财经大学 吴大军151 管理学 工商管理类 管理学 东北财经大学 卢昌崇152 管理学 工商管理类 中小企业管理 对外经济贸易大学 林汉川153 管理学 工商管理类 企业物流管理 湖南商学院 黄福华154 管理学 工商管理类 电子商务管理 华中师范大学 王学东155 管理学 工商管理类 基础会计学 暨南大学 宋献中156 管理学 工商管理类 管理学 辽宁大学 高闯号157 管理学 工商管理类 运营管理 南京大学 杨东涛158 管理学 工商管理类 创业管理 清华大学 雷家骕159 管理学 工商管理类 网络营销 山东大学 赵炳新160 管理学 工商管理类 管理学 武汉大学 谭力文161 管理学 工商管理类 电子商务概论 西安交通大学 李琪 162 管理学 工商管理类 审计学 中南财经政法大学 张龙平163 管理学 工商管理类 管理会计学 中央财经大学 孟焰 164 管理学 公共管理类 社区管理学 东北大学 孙萍 165 管理学 公共管理类 领导科学 吉林大学 彭向刚166 管理学 公共管理类 审计学 江西财经大学 张蕊 167 管理学 公共管理类 行政管理学 郑州大学 郑志龙168 管理学 公共管理类 社会保障学 中南财经政法大学 赵曼 169 管理学 公共管理类 行政伦理学 中南大学 李建华170 管理学 管理科学与工程类 生产管理 北京科技大学 张群 171 管理学 管理科学与工程类 信息系统分析与设计 合肥工业大学 梁昌勇172 管理学 管理科学与工程类 数据库系统及应用 江西财经大学 万常选173 管理学 管理科学与工程类 管理信息系统 清华大学 陈国青174 管理学 管理科学与工程类 生产计划与控制 上海交通大学 江志斌175 管理学 管理科学与工程类 运筹学 天津大学 吴育华176 管理学 管理科学与工程类 工程项目管理 同济大学 丁士昭177 管理学 管理科学与工程类 应用统计学 浙江大学 马庆国178 管理学 管理科学与工程类 管理学 中国地质大学(武汉)余敬 179 管理学 农业经济管理类 农产品营销学 华中农业大学 李崇光180 管理学 农业经济管理类 农产品运销学 南京农业大学 周应恒181 管理学 农业经济管理类 渔业经济学 上海水产大学 骆乐 182 管理学 图书档案学类 文书学 黑龙江大学 倪丽娟183 管理学 图书档案学类 信息检索与利用 山东理工大学 葛敬民184 管理学 图书档案学类 信息服务与用户 武汉大学 胡昌平185 管理学 图书档案学类 信息资源共享 中山大学 程焕文186 教育学 教育学类 教育技术学导论 北京师范大学 黄荣怀187 教育学 教育学类 教育技术学 河北大学 张立新188 教育学 教育学类 课程与教学论 湖南师范大学 石鸥 189 教育学 教育学类 职业教育学 天津工程师范学院 周明星190 教育学 教育学类 教育传播学 西北师范大学 杨改学191 教育学 教育学类 数学教育学 西南大学 宋乃庆192 教育学 体育学类 体育与健康 北京师范大学 毛振明193 教育学 体育学类 运动生物化学 广州体育学院 林文弢194 教育学 体育学类 学校体育学 华中师范大学 王健 195 教育学 体育学类 体育管理学 山东大学 张瑞林196 教育学 体育学类 人体解剖学 陕西师范大学 田振军197 教育学 体育学类 武术 上海体育学院 邱丕相号198 教育学 体育学类 篮球 苏州大学 王家宏199 教育学 体育学类 运动生理学 天津体育学院 张勇 200 教育学 体育学类 大学体育 西安交通大学 闫振龙201 经济学 经济学类 保险学原理 北京大学 孙祁祥202 经济学 经济学类 微观经济学 北京大学 于鸿君203 经济学 经济学类 货币银行学 东北财经大学 艾洪德204 经济学 经济学类 计量经济学 东北财经大学 王维国205 经济学 经济学类 商业银行管理学 湖南大学 彭建刚206 经济学 经济学类 统计学 南京财经大学 凌亢(凌迎兵)207 经济学 经济学类 国际金融学 南京大学 裴平 208 经济学 经济学类 西方经济学 南开大学 刘骏民209 经济学 经济学类 财政学 上海财经大学 蒋洪 210 经济学 经济学类 金融工程 武汉大学 叶永刚211 经济学 经济学类 政治经济学 西南财经大学 刘灿 212 经济学 经济学类 财政学 浙江财经学院 钟晓敏213 经济学 经济学类 国际贸易实务 浙江工业大学 程惠芳214 理学 材料科学类 材料化学导论 厦门大学 董炎明215 理学 大气科学类 数值天气预报 南京信息工程大学 陆维松216 理学 地理科学类 气象学与气候学 华东师范大学 束炯 217 理学 地理科学类 自然地理学 华南师范大学 徐颂军218 理学 地理科学类 GIS设计 南京大学 李满春219 理学 地理科学类 地理信息系统 首都师范大学 宫辉力220 理学 地球物理学类 地震学原理与应用 中国科学技术大学 刘斌 221 理学 地质学类 普通地质学 南京大学 舒良树222 理学 地质学类 岩石学 中国地质大学(武汉)桑隆康223 理学 地质学类 地史学 中国地质大学(武汉)龚一鸣224 理学 地质学类 沉积岩石学 中国石油大学(北京)朱筱敏225 理学 电子科学类 大学信息技术基础 湖南大学 李仁发226 理学 电子科学类 半导体器件物理与实验 吉林大学 孟庆巨227 理学 海洋科学类 声学基础 厦门大学 许肖梅228 理学 海洋科学类 卫星海洋学 中国海洋大学 刘玉光229 理学 化学类 物理化学 复旦大学 范康年230 理学 化学类 有机化学及实验 湖南大学 郭灿城231 理学 化学类 有机化学 华中农业大学 陈长水232 理学 化学类 分离科学 济南大学 罗川南233 理学 化学类 高分子物理学 青岛科技大学 吴其晔234 理学 化学类 仪器分析 清华大学 张新荣235 理学 化学类 绿色化学 四川大学 胡常伟236 理学 化学类 分析化学 武汉大学 张华山号237 理学 化学类 大学化学实验 武汉大学 席美云238 理学 化学类 无机化学与化学分析 西北大学 高胜利239 理学 化学类 物理化学 扬州大学 刁国旺240 理学 化学类 微型半微型有机化学实验 云南大学 李霁良241 理学 化学类 综合化学实验 浙江大学 王彦广242 理学 化学类 无机化学 中南大学 黄可龙243 理学 环境科学类 环境微生物学 浙江大学 郑平 244 理学 力学类 理论力学 湖南大学 刘又文245 理学 力学类 理论力学 兰州大学 周又和246 理学 生物科学类 植物生物学 北京师范大学 周云龙247 理学 生物科学类 城市生态学 海南大学 杨小波248 理学 生物科学类 生物信息学 华中农业大学 王石平249 理学 生物科学类 生态学 南京大学 安树青250 理学 生物科学类 基础生物学技术 南京大学 陈建群251 理学 生物科学类 微生物学 南开大学 李明春252 理学 生物科学类 生物化学 武汉大学 张楚富253 理学 生物科学类 动物生物学 厦门大学 陈小麟254 理学 生物科学类 动物生物学 云南大学 肖蘅 255 理学 数学类 数学分析 北京大学 彭立中256 理学 数学类 几何学及其习题 北京大学 莫小欢257 理学 数学类 数学建模 电子科技大学 黄廷祝258 理学 数学类 运筹学 东北电力大学 张杰 259 理学 数学类 数理统计 东北师范大学 史宁中260 理学 数学类 数学建模与数学实验 东南大学 朱道元261 理学 数学类 大学数学(农科) 华南农业大学 张国权262 理学 数学类 数学物理方程 吉林大学 尹景学263 理学 数学类 高等几何 南京师范大学 周兴和264 理学 数学类 微积分与数学实验 山东大学 吴臻 265 理学 数学类 常微分方程 四川大学 张伟年266 理学 数学类 实变函数与泛函分析 武汉大学 刘培德267 理学 数学类 高等代数 厦门大学 林亚南268 理学 数学类 线性代数与解析几何 中国科学技术大学 陈发来269 理学 天文学类 普通天文学 南京大学 李向东270 理学 统计学类 统计学 天津财经大学 肖红叶271 理学 统计学类 统计学 西南财经大学 向蓉美272 理学 物理学类 近代物理实验 北京大学 吴思诚273 理学 物理学类 核物理与粒子物理导论 北京大学 叶沿林274 理学 物理学类 大学物理实验 北京交通大学 成正维275 理学 物理学类 大学物理实验 福建师范大学 黄志高276 理学 物理学类 热力学与统计物理 复旦大学 孙鑫 277 理学 物理学类 中学物理课程与教学论 广西师范大学 罗星凯号278 理学 物理学类 光学 南京大学 丁剑平279 理学 物理学类 大学物理实验 南京理工大学 李相银280 理学 物理学类 大学物理实验 南开大学 高立模281 理学 物理学类 量子力学 清华大学 庄鹏飞282 理学 物理学类 原子物理学 山西大学 贾锁堂283 理学 物理学类 电磁学 中国科学技术大学 程福臻284 理学 心理学类 教育心理学 华东师范大学 吴庆麟285 理学 心理学类 教育心理学 华南师范大学 莫雷 286 历史学 历史学类 中国古代的政治与文化 北京大学 邓小南287 历史学 历史学类 中国少数民族史 兰州大学 王希隆288 历史学 历史学类 中国古代后期史 南开大学 李治安289 历史学 历史学类 区域社会史导论 山西大学 行龙 290 历史学 历史学类 世界近现代史 首都师范大学 徐蓝 291 历史学 历史学类 史前考古学 西北大学 张宏彦292 历史学 历史学类 中国古代史 厦门大学 陈支平293 两课 两课 毛泽东思想、邓小平理论和”三个代表“重要思想概论电子科技大学 邓淑华294 两课 两课 毛泽东思想、邓小平理论和“三个代表”重要思想概论福建农林大学 郑传芳295 两课 两课 民族理论与民族政策 广西民族大学 龚永辉296 两课 两课 思想道德修养与法律基础 湖南师范大学 李培超297 两课 两课 中国近现代史纲要 华中师范大学 刘伟 298 两课 两课 思想道德修养与法律基础 吉林大学 陈秉公299 农学 草业科学类 牧草及饲料作物育种学 内蒙古农业大学 云锦凤300 农学 动物生产类 家畜育种学 内蒙古农业大学 李金泉301 农学 动物医学类 家畜寄生虫学 广西大学 黄维义302 农学 动物医学类 动物微生物学 河南农业大学 崔保安303 农学 动物医学类 兽医药理学 华南农业大学 曾振灵304 农学 动物医学类 兽医内科学 华中农业大学 郭定宗305 农学 动物医学类 动物生物化学 南京农业大学 邹思湘306 农学 动物医学类 动物传染病学 四川农业大学 文心田307 农学 动物医学类 兽医免疫学 中国农业大学 杨汉春308 农学 环境生态类 土壤侵蚀原理 北京林业大学 张洪江309 农学 环境生态类 保护生物学 东北林业大学 迟德富310 农学 环境生态类 园林植物育种学 华中农业大学 包满珠311 农学 环境生态类 治沙原理与技术 内蒙古农业大学 高永 312 农学 环境生态类 农业生态学 山东农业大学 陈雨海313 农学 森林资源类 森林培育学 北京林业大学 马履一314 农学 森林资源类 园林树木学 华中农业大学 陈龙清315 农学 水产类 水生生物学 大连水产学院 赵文 316 农学 水产类 贝类增养殖学 中国海洋大学 王昭萍号317 农学 植物生产类 园艺植物生物技术 华中农业大学 邓秀新318 农学 植物生产类 生物统计与田间试验 南京农业大学 盖钧镒章元明319 农学 植物生产类 普通植物病理学 南京农业大学 刘凤权320 农学 植物生产类 作物育种学 石河子大学 曹连莆321 农学 植物生产类 普通植物病理学 云南农业大学 何月秋322 农学 植物生产类 普通昆虫学 中国农业大学 彩万志323 文化素质 文化素质教育课程类大学文化与成人之道 湖南大学 章兢324 文化素质 文化素质教育课程类中国女性文化 湖南师范大学 赵树勤325 文化素质 文化素质教育课程类数学文化 南开大学 顾沛326 文化素质 文化素质教育课程类国际关系分析 清华大学 阎学通327 文化素质 文化素质教育课程类中国审美文化史 山东大学 陈炎328 文化素质 文化素质教育课程类西方哲学智慧 中国人民大学 张志伟329 文化素质 文化素质教育课程类现代生命科学 中山大学 王金发330 文化素质 文化素质教育课程类非物质文化遗产概论 重庆文理学院 牟延林331 文学 外国语言文学类 大学英语视听说 北京理工大学 吴树敬332 文学 外国语言文学类 西班牙语口笔语实践(精读) 北京外国语大学 董燕生333 文学 外国语言文学类 大学英语 北京邮电大学 卢志鸿334 文学 外国语言文学类 高级商务英语听说 对外经济贸易大学 陈准民335 文学 外国语言文学类 阿拉伯语经贸谈判与口译 对外经济贸易大学 杨言洪336 文学 外国语言文学类 英语口译(课程系列) 广东外语外贸大学 仲伟合337 文学 外国语言文学类 大学英语 河北科技大学 张森 338 文学 外国语言文学类 高级英语 四川外语学院 肖肃 339 文学 外国语言文学类 基础日语 天津外国语学院 张晓希340 文学 外国语言文学类 大学德语 同济大学 朱建华341 文学 外国语言文学类 大学英语 西南交通大学 吕长竑342 文学 外国语言文学类 大学英语 湘潭大学 杨华 343 文学 外国语言文学类 基础朝鲜语(韩国语) 延边大学 金永寿344 文学 外国语言文学类 大学法语 中国海洋大学 李志清345 文学 外国语言文学类 综合俄语 中国人民解放军外国语学院郅友昌346 文学 外国语言文学类 大学英语 重庆大学 邹晓玲347 文学 新闻传播学类 新闻学概论 安徽大学 芮必峰。
物理化学(上)练习试题(二)一、选择题1. 一定量气体经历某一过程,体系的热力学能U 增加,则此过程 ( ) A. 是等压过程 B. 体系吸热 C. 体系温度升高 D. 是压缩过程2. 某气体的状态方程为m pV RT bp =+(常数b>0),此气体向真空吸热膨胀后,其浓度 ( )A. 上升B. 下降C. 不变D.不确定3. 关于状态函数,下列说法不正确的是 ( ) A. 状态函数的仍为状态函数 B. 状态函数的绝对值是不可测定的 C. 广度性质和强度性质都是状态函数 D. 状态函数的二阶可导数与求导顺序无关4. 下列公式不受理气条件限制的是 ( ) A.2ln W nRT V /V =−1T B. (K 为常数)11TV K ϒ=C. r m r m B, g H U R νΔ=Δ+∑D. ()()21r 2r m 1Bm d T p ,T H T H T C νΔ=Δ+∑∫T m5. 对于热力学能U 的意义,正确的是 ( ) A. 只有理气的热力学能是状态的单值函数 B. 对应于某一状态的热力学能是不可测定的 C. 当理气的状态改变时热力学能一定改变D. 体系的热力学能即为体系内分子之间的相互作用势能 6. 对于单组分相体系,下列物理量组中均为强度性质的是 ( ) A. T , U m , V B. m J T ,V ,H μ− C. m J T V ,,C ,U μ− D. m V V ,p,C 7. 热力学第一定律表示为 d U Q p V d =δ−,其中p 代表体系压力,该公式使用条件为 ( )A. 理气可逆过程B. 理气等压过程C. 封闭体系只作膨胀功的可逆过程D. 封闭体系恒外压过程 8. 下述正确的是 ( )A. 物体的温度越高,说明其热力学能U 越大B. 物体的温度越高,说明所含热量越多C. 体系温度升高,就肯定它吸收了热D. 体系温度不变,说明它既不吸热,也不放热9. 在同一高温热源与同一低温热源间工作的可逆热机其效率为R η,不可逆热机效率为IR η,则二者关系为 ( )A. R η= IR ηB. R η< IR ηC. R η> IR ηD. 不能确定 10. 对于可逆变化A → B ,有BRAQ S TδΔ=∫下述正确的是 ( )A. 只有可逆变化才有熵变B. 可逆变化没有热温熵C. 可逆变化熵变为零D. 可逆变化熵变与热温熵之和相等 11. 下列过程中体系的不等于零的是 ( )G ΔA. 液体苯在正常沸点汽化 B. 在273.2 K 常压下,水结成冰C. 373.2 K 大气上,水向真空蒸发成同温同压下的水蒸气D. 在大气中进行的反应 NH 4Cl (s) → NH 3 (g) + HCl (g) 12. 200 K 时,反应 CO (s) +12O 2 (g) === CO 2 (g) 的平衡常数,同温度下反应 CO K \2 (g) === 2 CO (g) + O 2 (g) 的平衡常数为 ( ) A. ()1/2p K \ B. 1Kp \C. ()1/21p K \D. ()21p K \13.,压力对平衡常数无影响,这句话适用的条件是 ( )B0ν=∑A. 高温高压下的反应体系 B. 理气反应体系C. 含有气相的高相反应体系D. 任何气体反应体系14. 在298 K ,101.3 kPa 下,反应C (s) + H 2 (g) ===12C 2H 4 (g),2 C (s) +3 H 2 (g)=== C 2H 6 (g),C 2H 4 (g) + H 2 (g) === C 2H 6 (g)的压力平衡常数分别为12,,p p p 3K K K ,则它们之间的关系为 ( )A. B.(1231/2/p p p K K K =)1p 2p 232/p p K K K =C. D. 1321/2p p p K K K =31p p K K K = 15. 二级组分系数最多可平衡共存的相数为 ( )A. 2B. 3C. 4D. 5 二、证明题1. 对范德华气体,证明22T U an V V∂⎛⎞=⎜⎟∂⎝⎠2. 对于理想气体体系,试证明体系压力p 是状态函数,而功W 不是状态函数。
600多个精品课程课件网络资源收录日期: 2007-03-16 (部分内容已严重陈旧或已淘汰)1 2005食品卫生检验教案讲稿2 2005最新国家食品卫生标准贯彻实施手册3 Catalysts for Fine Chemical Synthesis4 FDA农药分析手册5 FDA食品法规(2001版)6 Food Additives Date Book7 Food Outlook 英文版8 HACCP原理与实施第二版9 Instrumental Analysis10 Molecular Components of Cells11 WTO法律专题12 安徽建筑工程学院大学物理实验精品课程13 安徽建筑工程学院无机与分析化学精品课程14 安徽科技学院精品课程15 白兰地工艺学16 包装技术课件17 保健食品检验与评价技术规范实施手册18 保健食品原料手册19 贝类油脂化学与工艺学20 病理学实验指导-大连医科大学21 仓储与配送管理-天津开发区职业技术学院22 长春工业大学精品课程23 长春师范学院教学课件24 长效酸奶技术25 常见中毒急救手册26 常用分子生物学软件中文说明27 常用天然提取物质量标准参考手册200328 常用药物辅料手册29 成都大学精品课程建设30 成都中医药大学中药学精品课程31 成都中医药大学中药药理学精品课程32 成都中医药大学中医方剂学精品课程33 成都中医药大学中医药统计学精品课程34 大连水产学院大学.物理精品课程35 大连水产学院精品课程36 大连水产学院水产动物生理学精品课程37 大连水产学院水生生物学精品课程38 大连水产学院养殖水环境化学精品课程39 大学基础化学40 大学物理41 大学物理42 大学物理-西安科技大学43 蛋白质纯化与鉴定实验指南44 蛋白质电泳实验技术45 蛋白质电泳实验技术46 蛋白质技术手册47 蛋白质与氨基酸48 蛋白质组学-黎明涛49 蛋制品工艺学50 当代食品生产技术丛书-当代食品生产技术与配方51 当代食品生产技术丛书-调味品52 当代食品生产技术丛书-果蔬保鲜与加工53 当代食品生产技术丛书-酒类制造54 当代食品生产技术丛书-粮食深加工55 当代食品生产技术丛书-乳制品56 当代食品生产技术丛书-食用植物油与植物蛋白57 当代食品生产技术丛书-饮料58 当代中医药美容妙方59 电镜精品课件-江南大学60 电泳-生物化学实验技术丛书61 电子显微镜图像分析原理与应用62 调香术-化学工业出版社63 动物学64 动物学65 动物营养学66 毒理学67 发酵调味品生产技术68 发酵工程69 发酵工程70 发酵工艺及设备71 发酵工艺学课件72 发酵工艺学原理73 发酵食品工艺学74 发酵与酿造工艺学75 发酵原理课外讲座76 发酵原理演示教案77 方便食品加工工艺及设备选用手册78 方便食品原料学与工艺学79 分析化学80 分析化学81 分析化学82 分析化学83 分析化学电子教案84 分析化学讲义85 分子克隆实验指南-第三版86 分子生物学87 分子生物学88 分子生物学郜金荣89 分子生物学和食源性致病菌的快速检测90 分子生物学-特纳91 分子医学概论-周俊宜92 分子遗传学课件93 福州大学生物化学精品课程94 复旦大学精品课程95 概率与统计96 高级分子生物学要义97 个人空间98 各类手术病人的营养治疗99 各种香料英语电子书100 工业生物催化101 工业微生物学102 功能基因组学103 功能性色素在高新技术中的应用104 罐头工业手册105 广东工业大学精品课程106 广西师范大学精品课程107 广州体育学院运动生物化学精品课程108 国家基本药物临床手册(西药)109 果脯蜜饯生产工艺与配方110 果品蔬菜贮藏运销学111 果蔬食品加工工艺与配方112 果蔬贮运与加工-北京农业职业学院113 果树生产技术精品课程-新疆农业职业技术学院114 果树栽培-北京农业职业学院115 海水贝类增养殖学116 杭州师范学院动物学精品课程117 杭州师范学院动物学精品课程118 杭州师范学院精品课程119 杭州师范学院生理学精品课程120 合肥工业大学工程原理精品课程121 合肥工业大学精品课程122 河北大学动物生物学精品课程123 河北大学精品课程124 河北科技大学波谱解析精品课程125 河北科技大学化工原理精品课程126 河北科技大学化学反应工程精品课程127 河北科技大学精品课程128 河北科技大学生物化学精品课程129 河北科技大学微生物学精品课程130 河北科技大学细胞生物学精品课程131 河北科技大学质量管理学精品课程132 河北农业大学精品课程133 河南大学分析化学精品课程134 河南科技学院植物化学保护精品课程135 河南农业大学精品课程136 河南师范大学精品课程137 湖南大学分析化学在线教案138 湖南科技学院精品课程139 湖南农业大学精品课程140 湖南师范大学消费经济学精品课程141 护肤养颜中药(2003)142 护理学基础-哈尔滨医科大学143 护理学教案与课件-信阳职业技术学院144 护理学实验指导-信阳职业技术学院145 华南理工大学精品在线课程146 华南农业大学教学资源库147 华中科技大学环境卫生学精品课程148 华中科技大学外科学精品课程149 华中农业大学动物生理学精品课程150 华中农业大学基因操作原理精品课程151 华中农业大学生态学精品课程152 华中农业大学食品工程原理精品课程153 华中农业大学微生物生物学精品课程154 华中农业大学物理化学精品课程155 华中农业大学植物生理学精品课程156 华中农业大学植物营养学精品课程157 华中农业大学作物栽培学精品课程158 化工书籍159 化工原理160 化工原理161 化学反应工程162 环境工程微生物学163 环境生态学-西安科技大学164 环境卫生学165 黄冈职业技术学院-食品营养学166 基础化学-哈尔滨医科大学167 基础生物化学168 基础生物化学169 基因工程170 基因工程学原理171 吉林农业大学动物微生物学精品课程172 吉林农业大学精品课程173 即食食品中单增李斯特菌的风险评估174 集美大学精品课程175 技术检验学课件176 家传牛肉面配方大公开177 家禽生产-中央广播电视大学178 家庭保健药膳制作400法179 家庭养生百科全书180 检验技术181 江南大学食品工艺学精品课程182 江南大学食品化学精品课程183 江西农业大学精品课程184 江西农业大学微生物学精品课程185 结构化学186 精细化工产品配方合成及应用187 军队营养与食品卫生学188 抗癌动、植、矿物彩色图鉴及其应用189 抗生素工艺学190 矿泉水与纯净水工业手册191 莱阳农学院精品课程192 兰氏化学手册-第十三版中文版193 老年人实用营养知识194 冷冻食品加工技术与工艺配方195 历朝龙-生物化学与分子生物学实验技术196 粮油食品加工学197 -辽宁科学技术出版社198 临床微生物学199 临沂师范学院精品课程200 流体力学201 绿色食品生产工艺学-广东农工商职业技术学院202 酶-食品工业的魔术师203 美国现代食品科技系列204 美国现代食品科技系列-发酵食品微生物学205 美国现代食品科技系列-乳制品生产技术206 美国现代食品科技系列-食品化学207 美国药典208 美食营养金钥匙-西苑出版社209 免疫细胞的分离和保存技术210 面点制作211 膜技术手册-化学工业出版社212 默克家庭诊疗手册213 内蒙古大学大学物理精品课程214 内蒙古大学分析化学精品课程215 内蒙古大学基因工程原理精品课程216 内蒙古大学精品课程217 内蒙古大学普通生态学精品课程218 内蒙古大学生物化学精品课程219 内蒙古农业大学精品课程220 内蒙古农业大学植物学精品课程221 南方医科大学病原生物学精品课程222 南方医科大学内科学精品课程223 南京大学精品课程224 南京大学普通生物学精品课程225 南京大学物理化学精品课程226 南京理工大学精品课程227 南京农业大学精品课程228 酿造酒工艺学229 农产品加工工艺学230 农产品营销学231 农药残留检测技术232 农药中毒急救手册233 农业经济学234 农业推广技术235 欧盟动物源食品安全管理法规236 欧洲药典5.4237 烹饪技能全书238 品质漫画九千239 葡萄酒工艺学240 葡萄酒-湖南文艺出版社241 葡萄酒品尝学242 普通化学243 普通生物学244 普通生物学-生命科学通论245 普通微生物学教程246 普通微生物学-周德庆微生物学教程247 青岛科技大学精品课程248 清华大学仪器分析精品课程249 清华基因Gene课件250 全国中药炮制规范1988版251 全套仪器分析讲义252 染色与镜鉴253 肉类禽类产品微生物学254 乳品工艺学255 乳品和乳品加工技术256 乳品化学-郭本恒主编257 乳品加工手册258 乳品科学与技术精品课程259 乳与乳制品工艺学260 乳制品加工工艺261 山东农业大学-肉制品工艺学精品课程262 山东中医药大学中医基础理论精品课程263 山东中医药大学中医诊断学精品课程264 山西大学精品课程265 陕西师范大学精品课程266 陕西师范大学物理化学网络课堂267 陕西师范大学有机化学精品课程268 上海大学精品课程269 上海水产大学-食品加工学精品课程270 烧烤技术要诀271 神奇的乳酸菌-健康快车系列丛书272 沈阳农业大学精品课程273 生产技术274 生化分离技术275 生化与分子生物学课件276 生物化学277 生物化学278 生物化学279 生物化学280 生物化学281 生物化学-北京大学282 生物化学电子教案283 生物化学检验-湖北中医学院284 生物化学讲义285 生物化学教学讲义286 生物化学教学讲义287 生物化学课件288 生物化学实验指导-福建医科大学289 生物化学-西华大学290 生物化学与分子生物学技术-周俊宜291 生物化学与分子生物学实验技术292 生物化学-郑州牧业高等专科学校293 生物统计附实验设计294 生物信息学295 实验中医学-上海中医药大学296 实用安全手册297 实用烘烤食谱298 实用食物营养成分分析手册299 实用着色与配色技术300 食品安全301 食品安全质量鉴别与国家检验标准全书302 食品百科303 食品包装304 食品包装技术305 食品包装学-化学工业出版社306 食品保藏原理307 食品保藏原理308 食品标准309 食品的色香味艺术之色310 食品雕刻311 食品分析-河北科技大学312 食品分析技术-中国轻工业出版社313 食品分析-宁夏大学314 食品分析实验讲义-广东海洋大学315 食品分析实验-苏州农业职业技术学院316 食品分析实验指导-宁夏大学317 食品分析与感官评定-广东海洋大学318 食品风味学319 食品感官评价原理与技术320 食品工厂设计电子教案321 食品工程原理322 食品工程原理323 食品工业废水处理324 食品工艺学325 食品工艺学326 食品工艺学导论-面向二十一世纪系列教材327 食品工艺学-第二版328 食品工艺学-华中农业大学329 食品工艺学-江南大学330 食品工艺学实验指导-华中农业大学331 食品化学332 食品化学第四版-面向21世纪课程教材333 食品化学-高等职业教育教材334 食品化学-江南大学335 食品化学-苏州农业职业技术学院336 食品化学与分析337 食品化学与营养学338 食品化学与营养学339 食品化学-中国轻工业出版社340 食品机械与设备341 食品加工学实验342 食品加工与保藏原理343 食品检测技术344 食品抗氧化剂(实用篇)-经典英文原著345 食品科学-第三版346 食品课堂347 食品理化检测技术-广西职业技术学院348 食品理化与微生物检验实验349 食品论坛350 食品人才351 食品杀菌新技术352 食品生产技术353 食品生物化学354 食品生物化学声文并茂355 食品试验设计与统计分析356 食品添加剂电子书357 食品添加剂分析方法(英文原版)358 食品添加剂和污染物的评估359 食品添加剂实用手册360 食品添加剂手册361 食品添加剂制备工艺-广州科技出版社362 食品图库363 食品网刊364 食品网址365 食品微生物多媒体教程-食品伙伴网366 食品微生物学367 食品微生物学-广东海洋大学368 食品微生物学教程369 食品微生物学实验技术370 食品微生物学实验指导-广东海洋大学371 食品卫生常用法律法规手册372 食品卫生法-哈尔滨医科大学373 食品卫生学374 食品污染与健康-化学工业出版社375 食品营养与卫生学376 食品与环境生物技术377 食品与生活378 食品增味剂379 食品制作大全电子图书380 食品专题381 食品资讯382 食用菌生产与加工383 手性合成-不对称反应384 首页385 兽医微生物学386 兽医应用免疫学387 兽医应用免疫学388 蔬菜栽培-苏州农业职业技术学院389 双向电泳操作手册中文译本390 水产品加工技术391 水产养殖-中央广播电视大学392 四川理工学院精品课程393 四川理工学院食品保藏原理精品课程394 四川理工学院统计学精品课程395 四川理工学院微生物工程工艺原理精品课程396 四川理工学院无机化学精品课程397 四川理工学院仪器分析精品课程398 四川农业大学精品课程399 四川农业大学农业植物病理学-果蔬病害专题400 四川农业大学农业植物病理学-作物病害专题作者:foodmate2005401 饲料行业精品书籍16本402 苏州大学普通物理学精品课程403 速冻食品404 台湾大学-食品营养学概论405 台湾图书-多款肉丸子制作集锦406 天津科技大学包装结构设计精品课程407 天津科技大学精品课程408 天津农学院发酵微生物试验409 天津农学院基础微生物试验410 天津农学院仪器分析精品课程411 天津农学院遗传学精品课程412 天津商学院网上课堂413 天津师范大学网络学堂414 天然磷脂产品的加工及应用415 天然酸碱指示剂416 田间试验与统计分析417 统计学418 图片链接419 瓦楞纸箱生产实用技术420 外文微生物幻灯片421 微机在医学中的应用课件422 微生物快速检测技术的发展现状423 微生物生物技术-应用微生物学基础原理424 微生物实验室手册425 微生物实验室手册英文版426 微生物限量检查法427 微生物学428 微生物学429 微生物学430 微生物学431 微生物学432 微生物学辞典433 微生物学和微生物学检验434 微生物学检验-信阳职业技术学院435 微生物学教程436 微生物学教程-周德庆编著437 微生物学-沈萍438 微生物学实验指导439 微生物资源开发利用440 维生素手册441 潍坊职业学院-食用菌栽培442 卫生统计学443 无机材料物理化学444 无机化学445 无机化学446 无机化学447 无机化学-哈尔滨医科大学448 无机化学教案449 无机化学与分析化学450 无机及分析化学451 无机及分析化学452 无机与分析化学-苏州农业职业技术学院453 无菌检查法演示实验454 武汉大学精品课程455 武汉大学普通物理实验456 武汉大学微生物专题网站457 武汉大学药理学精品课程458 武汉工业学院精品课程459 物理化学460 物理化学461 物理化学462 物理化学463 物理化学课件464 物理化学-西安科技大学465 物流学概论-北京物资学院466 物质的颜色与结构-北京师范大学出版社467 西北大学化工原理精品课程468 西北大学微生物学精品课程469 西北农林科技大学精品课程470 西北农林科技大学畜产食品工艺学精品课程471 西昌学院动物微生物学及免疫学精品课程472 西昌学院食品工程原理精品课程473 西昌学院食品生物化学精品课程474 西昌学院畜牧生产学精品课程475 西昌学院植物生理学精品课程476 西昌学院作物栽培学精品课程477 西华大学精品课程478 西华大学食品工艺学精品课程479 西华大学微生物学精品课程480 西南农业大学精品课程481 西南石油学院大学物理与实验精品课程482 西南石油学院化工原理精品课程483 细胞生物学484 细胞生物学485 细胞微生物学486 厦门大学精品课程487 厦门大学精品课件488 现场改善489 现代保健食品研发与生产新技术490 现代冻干技术491 现代分子生物学-第二版492 现代科学技术493 现代农业技术专题讲座494 现代企业管理方法495 现代生物实验教案496 现代生物与食品技术497 现代食品安全检测技术498 现代食品微生物学-中国轻工业出版社499 现代微生物学500 现代微生物学与实验技术-科学出版社作者:foodmate2005501 现代营养学(原著第八版)502 香料化学与工艺学503 湘潭大学大学化学基础精品课程504 消毒技术规范2002年版505 新版面包配方-中国轻工业出版社506 新编分子生物学实验指南507 新编中药志508 新编中药志(三)509 新编中药志(四)510 新疆石河子大学-水处理微生物学精品课程511 新型食源性致病菌检测-显色培养基512 性药学513 徐州医学院生理学精品课程514 徐州医学院生物化学与分子生物学精品课程515 畜产食品工艺学516 养牛生产-中央广播电视大学517 养生集-一部让中国人多活十年的书518 养兔生产-中央广播电视大学519 养羊生产-中央广播电视大学520 养殖水域生态学521 养猪生产-中央广播电视大学522 样品处理技术523 药材标准品种大全524 药品注册工作手册525 药品注册指南526 药物化学527 药物研究中的分子识别528 药用辅料手册第四版529 药用辅料应用技术第二版2002530 药用辅料质量管理规范与现代辅料新技术应用全书531 医学本草纲目电子版532 医学电子学课件533 医学微生物学534 医学微生物学535 仪器分析536 仪器分析537 仪器分析538 仪器分析-河北科技大学539 仪器分析化学540 仪器设备541 遗传学课件542 益生菌基础与应用543 饮料的配方与工艺544 饮料技术手册545 饮食疗方1500例546 饮用水与寄生虫547 应用微生物学实验法548 应用微生物学实验法549 英语食品电子书550 营养与食品卫生学551 优良花卉品种及其栽培技术-中央广播电视大学552 优良蔬菜品种及其栽培技术-中央广播电视大学553 油炸食品配方及制作方法554 有关食品设备的精品书籍555 有机化学556 有机化学557 有机化学558 有机化学559 有机化学-哈尔滨医科大学560 有机化学实验-北大版561 有机化学实验-清华版562 有机化学实验-上海交大版563 有机化学实验-武汉大学版564 有机化学-西安科技大学565 有机实验视频课件566 鱼类学567 鱼类学-广东海洋大学568 原子吸收在环境分析中的应用569 原子吸收在卫生检验中的应用570 云南大学食品化学精品课程571 枣庄学院大学物理精品课程572 枣庄学院动物生物学精品课程573 枣庄学院精品课程574 枣庄学院植物生物学精品课程575 藻类植物实验576 浙江财经学院精品课程577 浙江大学保护生物学精品课程578 浙江大学动物学精品课程579 浙江大学普通生物学精品课程580 浙江大学神经生物学精品课程581 浙江大学生态学精品课程582 浙江大学生物化学精品课程583 浙江大学生物物理学精品课程584 浙江大学生物信息学精品课程585 浙江大学细胞工程精品课程586 浙江大学细胞生物学精品课程587 浙江大学遗传学精品课程588 浙江大学植物生理学精品课程589 浙江大学植物学精品课程590 浙江林学院精品课程591 浙江医药高等专科学校药剂学精品课程592 浙江医药高等专科学校医药基础课件593 正交与均匀实验设计594 郑州牧业高等专科学校教学网上资源595 郑州牧业高等专科学校精品课程596 郑州牧专动物解剖学与组织胚胎学597 郑州牧专生物化学精品课程598 郑州牧专细胞生物学及细胞培养精品课程599 郑州轻工业学院精品课程600 政策法规作者:foodmate2005601 植物生物学602 植物水势、溶质势和叶绿素含量测定实验603 植物组织培养技术604 制冷与技术课件605 质量工程师手册(上)606 质量工程师手册(下)607 质量管理608 质量管理的精品书籍609 质量检验教程610 中国科技大学大学物理实验精品课程611 中国科技大学定量分析化学精品课程612 中国科技大学精品课程613 中国科技大学生理学网络课件614 中国科技大学无机化学精品课程615 中国科技大学有机化学精品课程616 中国美食丛书-中国野菜食谱大全617 中国药茶618 中国药典2005版第三部619 中国药品检验标准操作规范620 中国药品检验标准操作规范与药品检验仪器操作规程2005年版大全621 中国饮食营养学-北京中医药大学622 中国中药材真伪鉴别图典(特经典)623 中华人民共和国兽药典第一部2000624 中华食品工业大辞典625 中药保健食品研制和开发626 中药名大全627 中药新药临床研究指导原则628 中药学鉴定-湖北中医学院629 中药制剂检验技术-山西生物应用职业技术学院630 中药制药工艺技术解析631 种子产业化技术-苏州农业职业技术学院632 专业英语633 资料中心634 综合实验指导635 最新食品工业生产新工艺新技术与创新配方设计及产品分析检验实用手册636 最新食品添加剂品种优化选择与性能分析检测标准及应用工艺实用手册637 作物栽培学-宁夏大学。
陕西省教育厅关于公布2012年度改造升级省级精品课
程名单的通知
文章属性
•【制定机关】陕西省教育厅
•【公布日期】2012.12.03
•【字号】陕教高[2012]37号
•【施行日期】2012.12.03
•【效力等级】地方规范性文件
•【时效性】现行有效
•【主题分类】高等教育
正文
陕西省教育厅关于公布2012年度改造升级省级精品课程名单
的通知
(陕教高〔2012〕37号)
各普通高等学校:
按照《关于做好2012年度省级专业综合改革试点项目建设内容细化及遴选工作的通知》(陕教高办〔2012〕23号)精神和要求,省教育厅组织专家对各高校上报的已改造升级的原省级精品课程进行了评审。
省教育厅研究,确定西安交通大学的《材料科学基础》等100门课程为省级精品课程(名单见附件),同时,列入省级精品资源共享课建设行列,特提出建设要求如下:
一、列入2012年省级精品资源共享课建设计划的课程,需按照精品资源共享课的定位和要求(参见教高厅〔2012〕2号文《精品资源共享课建设工作实施办法》)进行建设。
二、各高校需对列入精品资源共享课建设的课程进行组织和制作,实行严格的课程内容审查制度。
三、各高校要及时将精品课程的建设情况反馈给省教育厅高等教育处,并在
一年内完成课程的建设任务,由省教育厅组织专家对精品课程的建设情况进行检查。
陕西省教育厅
2012年12月3日附件。
158 Univ. Chem. 2023, 38 (6), 158–163收稿:2023-02-27;录用:2023-05-08;网络发表:2023-05-15*通讯作者,Email:***************.cn基金资助:教育部产学合作协调育人项目(221004*********)•专题• doi: 10.3866/PKU.DXHX202302064 基于高阶思维能力的物理化学STS 习题设计与实践——以“热力学基本定律”为例陈亚芍*,冯佳米,王长号,张颖,白翠娥陕西师范大学化学化工学院,西安 710119摘要:在高等教育高质量、现代化发展的趋势下,对学生高阶思维能力的培养成为高等教育发展的重要方向。
本文在对高阶思维能力与STS (Science, Technology, Society)教育内涵理解的基础上,梳理STS 习题中实物条件、数量条件、特征条件、闲置条件等已知条件与高阶思维能力培养的关系,设计物理化学STS 习题。
以热力学基本定律为例,结合教学目标、学情、高阶思维能力培养的STS 习题设计规划,建立其知识点与STS 教育结合的双向明细表,考虑习题题型和难度,采用习题的筛选、改编、自编形式进行STS 习题设计,并应用于物理化学教学实践,促使学生高阶思维能力的提升,为物理化学习题内容、形式的改进提供了新思路。
关键词:高阶思维能力;STS 习题设计与实践;热力学基本定律中图分类号:G642.1;O6Design and Practice of Physical Chemistry STS Exercises toCultivate Higher-Order Thinking Skills: “Fundamental Laws of Thermodynamics” as an ExampleYashao Chen *, Jiami Feng, Changhao Wang, Ying Zhang, Cuie BaiCollege of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710119, China.Abstract: With the emerging trend of high-quality and modern higher education, cultivating higher-order thinking skills among students has become an important strategy for higher education development. Based on our understanding of the connotation of higher-order thinking skills and STS (science, technology, and society) education, this study organized the known conditions in STS exercises using the relationship between physical, quantitative, characteristic, and idle conditions and the cultivation of higher-order thinking skills. Considering the fundamental laws of thermodynamics in physical chemistry as an example, a two-way list of knowledge points combined with STS education was established by integrating teaching objectives, learning situations, and STS exercise planning, which was conducive to higher-order thinking skill cultivation. Considering the type and difficulty of STS exercises, they were designed in the form of selection, adaptation, and self-compilation and practically applied in physical chemistry teaching. These STS exercises promoted the higher-order thinking skills of students via problem solving. Thus, this study provides a novel approach to improve the content and forms of physical chemistry exercises.Key Words: Higher-order thinking skills; Design and practice of STS exercise;Fundamental laws of thermodynamics为推进高等教育的高质量发展,教育部提出“两性一度”的课程建设标准,将学生高阶思维能No. 6doi: 10.3866/PKU.DXHX202302064 159力的培养作为高等教育发展的重要方向。
Pure Appl. Chem., Vol. 72, No. 7, pp. 1357–1363, 2000.© 2000 IUP AC1357*Pure Appl. Chem. V ol. 72, No. 7, 2000. A special topic issue on green chemistry.Frontiers in green chemistry utilizing carbondioxide for polymer synthesis and applications*Jennifer L. Y oung and Joseph M. DeSimoneDepartment of Chemistry, Venable and Kenan Laboratories, University of NorthCarolina, Chapel Hill, NC 27599-3290 USAAbstract : In the last ten years, there has been incredible growth in research involving the use of carbon dioxide as an environmentally benign solvent. This article will highlight polymer synthesis, characterization, and applications in CO 2 and place this research in the context of both green chemistry and the latest developments in CO 2 technology.INTRODUCTIONIn recent years, the use of liquid and supercritical CO 2 as a green, environmentally benign solvent for chemical reactions and polymerizations has become a widespread, growing reality, both in academia and in industry [1–5]. CO 2 is a nontoxic, nonflammable, and inexpensive solvent. Although CO 2 is a “greenhouse gas”, CO 2 can be acquired from natural reservoirs or recovered as a by-product of industrial chemical processes, so no new production of CO 2 is necessary and there will be no additional contribution to the greenhouse gases. While CO 2 is a gas at ambient conditions, its liquid and supercritical states are easily attained by compression and heat. The critical conditions of 31.1 °C and 73.8 bar are relatively low. Liquid CO 2 is a compressible fluid, while supercritical CO 2 has relatively high liquid-like densities and low gas-like viscosities. Both liquid and supercritical CO 2 have a tuneable density (and dielectric constant) that increases with increasing pressure or decreasing temperature [6]. The solvency of supercritical CO 2 is often compared to that of fluorocarbon solvents. Many small molecules are soluble in CO 2 [7], including high vapor pressure solvents like methanol, acetone, and tetrahydrofuran, many vinyl monomers, and azo- and peroxy-initiators. Water and ionic compounds are insoluble [8,9], as are most polymers. Only two classes of polymers are soluble in CO 2 at relatively mild conditions (T < 100 °C, P < 350 bar), amorphous or low-melting fluoropolymers and silicone-based polymers[10]. Herein, polymer synthesis, characterization, and applications in CO 2 will be described, highlighting the environmental benefits of using CO 2.POL YMER SYNTHESISIn CO 2, few polymers, typically amorphous fluoropolymers, can be synthesized by a homogeneous,solution polymerization. Many insoluble polymers can be synthesized by a heterogeneous chain-growth process, such as precipitation, emulsion, dispersion, or suspension. Due to the solubility of many vinyl monomers and free-radical initiators in CO 2 and the ability to design appropriate CO 2-soluble polymeric surfactants, dispersion polymerization is a common heterogeneous polymerization method in CO 2. Step-growth polymerizations can also be conducted in CO 2 with advantages over other processes. Most of the polymers synthesized in CO 2 to date can be classified into three categories: fluoropolymers, dispersion polymers, and step-growth polymers.陕西师范大学物理化学精品课程1358J. L. YOUNG AND J. M. DESIMONE© 2000 IUP AC, Pure and Applied Chemistry 72, 1357–1363FluoropolymersFluoropolymer synthesis in CO 2, which is the subject of a recent chapter review [11], will now be discussed in the context of green chemistry. The synthesis of fluoropolymers in CO 2 is environmentally advantageous since these polymers are typically prepared in chlorofluorocarbons (CFCs) and other fluorinated solvents as well as in water. The association of CFCs with ozone layer destruction and the environmentally harmful nature and expense of fluorinated solvents necessitate the search for replacement solvents. Many fluoropolymerizations cannot be conducted in hydrocarbon solvents because of the detrimental side reactions arising from radical abstraction of hydrogen atoms from the solvent. Since CO 2 has no hydrogens to abstract, there is no chain transfer to CO 2. Amorphous fluoropolymers, such as highly fluorinated (meth)acrylates and perfluoropolyethers, are highly soluble in CO 2 and have been synthesized by solution polymerizations in supercritical CO 2 [12,13]. Low-molecular-weight oligomers of poly(tetrafluoroethylene) (PTFE) and poly(1,1-difluoroethylene) (PVDF), which remain soluble in CO 2 below a limiting molecular weight, have been synthesized by telomerizations in CO 2 [14,15]. Semi-crystalline, high-melting-temperature fluoropolymers, such as PTFE and PVDF, are insoluble in CO 2due to their crystallinity and are polymerized by various heterogeneous methods in CO 2 [16–18].Fluorinated acrylate polymers have been synthesized by homogeneous, solution polymerizations in CO 2 [12]. Using azobis(isobutyronitrile) (AIBN) as the thermal free-radical initiator, 1,1-dihydroperfluorooctyl acrylate (FOA) was polymerized at 60 °C, 207 bar. The high-molecular-weight polymer, PFOA, remained soluble in supercritical CO 2 and was identical to that polymerized in a CFC.It was also shown that statistical copolymers containing FOA and either methyl methacrylate, styrene,butyl acrylate, or ethylene, could be polymerized under homogeneous, solution polymerization condi-tions in CO 2. PFOA and CO 2-soluble copolymers containing PFOA are useful polymeric surfactants for applications including dispersion polymerizations and extractions, as will be shown later in this review.Perfluoropolyethers, which find applications as high-performance lubricants and heat-transfer fluids, constitute another class of fluoropolymers that have been synthesized in CO 2 [13]. The photooxi-dation of hexafluoropropylene (HFP) was performed in liquid CO 2 at –40 °C to yield perfluoropolyethers soluble in CO 2. This study showed that CO 2 is a viable solvent replacement for the dichlorodifluo-romethane currently used in some commercial fluoroolefin photooxidations.Tetrafluoroethylene (TFE) polymerizations in CO 2 are sensible because this dangerous, poten-tially explosive monomer is safely stored and handled as a pseudo-azeotrope with CO 2 [19,20]. Copo-lymerizations of TFE with perfluoro(propyl vinyl ether) (PPVE) or HFP in CO 2 have produced poly-mers with levels of comonomer incorporation similar to commercial polymers [16]. The TFE-PPVE copolymers also contained an unprecedented 0–3 acid endgroups per 106 carbon atoms, which are comparable to copolymers synthesized in CFCs and treated with a harsh fluorination process. TFE was also polymerized to high molecular weights in a biphasic mixture of CO 2 and water by heterogeneous methods similar to the industrial suspension and dispersion processes [17]. Using a persulfate initiator in the absence of a surfactant, aggregates with diameters of 0.5–3 m m were produced and resemble the granular PTFE formed commercially by suspension polymerization in water. In the presence of a CO 2-soluble surfactant, particles with diameters of 100–200 nm were produced and are similar to those made commercially by conventional aqueous dispersion polymerization.In the first continuous polymerizations in CO 2 reported recently [18], PVDF and poly(acrylic acid) were produced by continuous precipitation polymerization. A continuous polymerization process is much more commercially and environmentally viable than a batch polymerization, producing a larger amount of polymer in a given amount of time. The PVDF was formed at 75 °C, 275 bar and collected in a filter after polymerization residence times of between 15 and 40 min in the continuous stirred tank reactor.陕西师范大学物理化学精品课程Green chemistry utilizing carbon dioxide for polymers1359© 2000 IUP AC, Pure and Applied Chemistry 72, 1357–1363Dispersion polymers CO 2 is well suited as a reaction medium for dispersion polymerizations because many vinyl monomers and polymerization initiators are soluble in CO 2 while the polymers formed are insoluble [1,21].Surfactants for steric stabilization of the colloidal polymer particles in CO 2 must be soluble and must adsorb or chemically graft to the polymer particles and can consist of homopolymers, block, graft, and random copolymers, and reactive macromonomers. Polymer latexes produced in CO 2 eliminate volatile organic compound (VOC) emissions and can significantly reduce the energy-intensive drying process since CO 2 has a heat of vaporization which is low in the liquid state and zero in the supercritical state.Furthermore, polymer can be shipped dry as 100% solids and later be redispersed to save energy and money in shipping rather than shipping a heavy water latex.Initial studies illustrated the dispersion polymerization of methyl methacrylate (MMA) using PFOA as the stabilizer [22,23]. The next study of MMA dispersion polymerizations focused on the use of another class of surfactants, based on poly(dimethyl siloxane) (PDMS), which are less expensive than fluoropolymer-based surfactants [24]. Initial attempts of styrene dispersion polymerizations in CO 2 with PFOA and PDMS homopolymer stabilizers gave low yields and latex instability, but the block copolymers PS-b -PFOA and PS-b -PDMS produced submicron-sized PS particles [25,26]. Block co-polymer surfactants containing poly(vinyl acetate) (PV Ac), PV Ac-b -PDMS and PV Ac-b -PFOA, were effective stabilizers for dispersion polymerizations of vinyl acetate and copolymerizations of vinyl ac-etate and ethylene [27]. These dispersion polymerizations are summarized more extensively in a recent review with other examples [1].Recently, PFOA and its methacrylate analog PFOMA were found to be effective stabilizers for PS at higher CO 2 pressures (370 bar) [28] while PFOA was ineffective at lower pressures of 204 bar [25].Random copolymers, PS-co -PFOMA and PS-co -TMSOSPMA (3-[tris(trimethylsilyloxy)silyl]propyl methacrylate), are also effective stabilizers, producing micron-sized PS particles [29]. Such random copolymer surfactants, which can be prepared by a free-radical polymerization, are easier to synthesize than block copolymer surfactants, which must be prepared by “controlled” radical polymerization or living anionic polymerization. Recently, several other monomers have been polymerized by dispersion polymerizations, including 1-vinyl-2-pyrrolidone [30], acrylonitrile [31], hydroxyethyl methacrylate[32], and glycidyl methacrylate [33]. These examples extend the dispersion polymerizations into water-soluble, polar polymers and functionalized polymers.A new concept for the preparation of water-dispersible polymer particles in CO 2 has been devel-oped by Johnston [34]. PMMA was synthesized in CO 2 using an “ambidextrous” surfactant. The surfac-tant contains a water-soluble polymer block that anchors to the particle in CO 2 but becomes the electro-static stabilizer in water and a PDMS block that is the steric stabilizer in CO 2 but collapses and anchors to the particle in water. The PDMS-b -poly(methacrylic acid) surfactant was less effective during the dispersion polymerization of MMA in CO 2, but up to 10 wt% of the PMMA particles formed could be redispersed in water at pH 8 or 11. The other surfactant, PDMS-g -poly(pyrrolidonecarboxylic acid) was more effective during the dispersion polymerization, but the PMMA particles formed could not be redispersed in water.A recent report demonstrates the synthesis of highly cross-linked polymer particles in supercritical CO 2 [35]. Even in the absence of a stabilizer, the polymerization of divinylbenzene and ethylvinylbenzene produced polymer particles 1–5 m m in diameter. Addition of a block copolymer containing PMMA and a fluorinated polymethacrylate produced submicron-sized, monodisperse particles. Two other cross-linking monomers, ethylene glycol dimethacrylate (EGDM) and trimethylolpropane trimethacrylate (TRM) formed agglomerates of particles when polymerized in the absence of a stabilizer. At higher EGDM and TRM monomer concentrations (40–60%), continuous macroporous polymer monoliths were produced and pore diameters could be varied from 7880 to 20 nm by varying the monomer con-centration and CO 2 pressure [36].陕西师范大学物理化学精品课程1360J. L. YOUNG AND J. M. DESIMONE© 2000 IUP AC, Pure and Applied Chemistry 72, 1357–1363The chain-growth polymerizations discussed so far in this paper have been free-radical polymer-izations, typically initiated by AIBN. Using “controlled” free-radical polymerization techniques, such as atom transfer radical polymerization (A TRP), it is possible to obtain polymer chains with a very narrow molecular weight distribution and to synthesize block copolymers. Using Cu(0), CuCl, a functionalized bipyridine ligand, and an alkyl halide initiator, the block copolymers containing PMMA or PDMAEMA (DMAEMA = 2-(dimethylamino)ethyl methacrylate) were synthesized in CO 2 [37].Also in this report the dispersion polymerization in CO 2 of MMA with PFOA as the stabilizer was conducted by A TRP . The resulting PMMA had a relatively narrow molecular weight distribution (M w /M n = 1.4) in comparison to the same dispersion polymerization initiated by AIBN (M w /M n = 2-3) [23].Step-growth polymersThe recent advances in step-growth polymerizations in CO 2 have focused on poly(bisphenol A carbonate).One of the two primary industrial methods for polycarbonate synthesis uses methylene chloride, which is environmentally unfriendly, and phosgene, which is highly toxic. The other method, melt-phase polymerization, uses no solvent, but is limited by the high polymer melt viscosities and the high temperatures necessary for polymerization and processing. The presence of CO 2 in a melt polymerization plasticizes the polymer and reduces the viscosity, so the condensate can be removed more easily and higher molecular weight polymer can be achieved. The condensate, phenol, is soluble in sc CO 2 and can be efficiently extracted from the polymer by supercritical fluid extraction. Low-molecular-weight polycarbonate was shown to swell up to 55% at 235 °C, 350 bar [38]. Also, in this study, bisphenol A and diphenyl carbonate were polymerized to produce molecular weights up to 13 kg/mol. Under each reaction conditions, higher molecular weights were achieved by sc CO 2 extraction as opposed to phenol removal by argon.Solid-state polymerization of polycarbonate has also been studied using sc CO 2 to crystallize the polymer and to remove the phenol condensate [39,40]. In a solid-state polymerization, low-molecular-weight, semi-crystalline step-growth polymer beads are heated between the polymer Tg and Tm to allow for condensate and chain mobility for further chain extension reactions. Following the crystalliza-tion of beads of low-molecular-weight polycarbonate prepolymer by sc CO 2, solid-state polymeriza-tions were conducted. The polymerization rates in the solid state were found to be always greater with condensate removal facilitated by sc CO 2 as opposed to nitrogen under comparable conditions. Also,since the Tg of polycarbonate is decreased by over 70 °C in the presence of sc CO 2, the polymerization can be run at much lower temperatures, saving energy and suppressing the side reactions that lead to unwanted polymer discoloration.CHARACTERIZATIONIn order to study polymers and polymerization mechanisms in sc CO 2, analytical techniques have been modified for high pressure. Methods including IR, UV-vis, and NMR spectroscopies and scattering techniques are now possible at high pressures. This paper will focus on scattering techniques, including small-angle neutron scattering (SANS), small-angle X-ray scattering (SAXS), and light scattering.Both SANS and SAXS are powerful tools for determining the polymer radius of gyration, the second virial coefficient, A 2, which indicates the solvent quality for the polymer, and block copolymer micelle formation. SANS studies have shown that CO 2 is a good solvent for PFOA, a theta solvent for poly(hexafluoropropylene oxide), and a poor solvent for PDMS [41,42]. More recently, SANS research has shown a temperature- and pressure-induced theta point for PDMS in CO 2 [43]. At conditions above the theta point, CO 2 becomes a good solvent for PDMS. The aggregation behavior of three different amphiphiles in CO 2 was studied by SAXS [44]. The graft copolymer with a PFOA backbone and poly(ethylene oxide) (PEO) grafts formed micelles which stabilized water in CO 2, while the PTFE-b -PEO copolymer formed reverse micelles, and F(CF 2)10(CH 2)10H formed small aggregates of at most陕西师范大学物理化学精品课程Green chemistry utilizing carbon dioxide for polymers 1361© 2000 IUP AC, Pure and Applied Chemistry 72, 1357–1363four chains. The block copolymer PS-b -PFOA forms micelles in CO 2 with a PS core and a PFOA shell and the data fits a polydisperse spherical core-shell model [45,46]. With an increase in CO 2 density, the change in polydispersity suggests the existence of a critical micelle density, or a density of CO 2 above which the micelles are broken up into soluble unimers.High pressure static and dynamic light scattering were used to confirm the existence of a critical micelle density for block copolymers of PV Ac and poly(1,1,2,2-tetrahydroperfluoroalkyl acrylate)(PTAN) [47]. The block copolymer is insoluble at low CO 2 densities, forms spherical micelles at inter-mediate densities, and exists as unimers, or free polymer chains in solution, at high CO 2 densities. The transition of micelles-to-unimers was found to be very sharp, with the aggregation number decreasing from 120 to 1 with an increase in density.The in situ analysis of dispersion polymerizations in CO 2 has provided insight into the mecha-nism of the polymerizations. Turbidimetry, tensiometry, and dynamic light scattering confirmed that the critical flocculation density occurred at the theta point of the surfactant for poly(2-ethylhexyl acrylate)dispersed and sterically stabilized in CO 2 [48,49]. Turbidimetry was also used to study the dispersion polymerizations of vinyl acetate [27]. This in situ technique for measuring the polymer particle size is particularly useful since the PV Ac particle shape is lost during the removal of CO 2 due to the low Tg of the polymer. The dispersion polymerization of MMA stabilized by PDMS-monomethacrylate has also been the subject of mechanistic investigations [50,51]. The particle formation mechanism, studied by turbidimetry, involved coagulative nucleation and controlled coagulation [51]. The results obtained during the particle formation and particle growth stages agreed with the previous experimental study of Shaffer et al . [24].APPLICATIONS AND OUTLOOKWith the design of specific polymers as surfactants for CO 2, numerous environmentally benign applications for this clean, green solvent can be realized, of which extractions, cleaning, and coatings are just a few examples. For extractions and cleaning, the surfactant acts as a detergent to bring the insoluble species or contaminant into the CO 2 phase. Depressurization of the CO 2 or changing the CO 2density will cause the contaminant to be released from the surfactant. For example, a block copolymer was used to extract deuterated oligomers into CO 2 [45], and a dendrimer was used to extract a water soluble, CO 2-insoluble dye from the water phase into the CO 2 phase [52]. Detergents for CO 2 can be used for cleaning applications, such as “dry cleaning” using liquid CO 2 as the solvent instead of the common perchloroethylene, a probable human carcinogen [53]. In coating, the polymer to be delivered from the CO 2 can be soluble, such as a fluoropolymer, or an insoluble dispersed species. The application of coatings from CO 2 has been demonstrated by three different methods, including spray, spin, and free-meniscus coating, and extends CO 2 into areas such as microlithography, hard disk lubrication, and protective coatings [54]. Clearly, CO 2 can be used widely as an environmentally benign solvent for polymer synthesis and for numerous polymer-based applications.REFERENCES1.J. L. Kendall, D. A. Canelas, J. L.Y oung, J. M. DeSimone. Chem. Rev. 99, 543–563 (1999).2.P . G. Jessop and W. Leitner. Chemical Synthesis Using Supercritical Fluids , Wiley-VCH, Weinheim;Chichester (1999).3.W. Leitner. In Modern Solvents in Organic Chemistry , pp 107–132, Springer-V erlag, Berlin (1999).4.A. I. Cooper. J. Mater . Chem. 10, 207–234 (2000).5.J. M. DeSimone, W. Tumas, J. L. Y oung, P . A. Anastas. Green Chemistry Using Liquid and Supercritical Carbon Dioxide , Oxford University Press, Oxford, submitted for publication (2000).6. F. G. Keyes and J. G. Kirkwood. Phys. Rev. 36, 754 (1930).陕西师范大学物理化学精品课程1362J. L. YOUNG AND J. M. DESIMONE© 2000 IUP AC, Pure and Applied Chemistry 72, 1357–13637.J. A. Hyatt. J. Org. Chem. 49, 5097–5101 (1984).8.H. H. Lowry and W. R. Erickson. J. Am. Chem. Soc. 49, 2729 (1927).9.M. B. King, A. Mubarak, J. D. Kim, T. R. Bott. J. Supercrit. Fluids 5, 296 (1992).10.M. A. McHugh and V . J. Krukonis. Supercritical Fluids Extraction: Principles and Practice ; 2nd ed.; Butterworth-Heinemann, Boston (1994).11.J. P . DeY oung, T. J. Romack, J. M. DeSimone. In Fluoropolymers 1: Synthesis , Hougham, et. al.,(Eds.), pp 191–205. Plenum Press, New Y ork (1999).12.J. M. DeSimone, Z. Guan, C. S. Eisbernd. Science 257, 945–947 (1992).13.W. C. Bunyard, T. J. Romack, J. M. DeSimone. Macromolecules , 32 8224–8226 (1999).14.T. J. Romack, J. R. Combes, J. M. DeSimone. Macromolecules 28, 1724–1726 (1995).15.J. R. Combes, Z. Guan, J. M. DeSimone. Macromolecules 27, 865–867 (1994).16.T. J. Romack, J. M. DeSimone, T. A. Treat. Macromolecules 28, 8429–8431 (1995).17.T. J. Romack, B. E. Kipp, J. M. DeSimone. Macromolecules 28, 8432–8434 (1995).18.P . A. Charpentier, K. A. Kennedy, J. M. DeSimone, G. W. Roberts. Macromolecules 32, 5973–5975 (1999).19.S. V . Gangal. In Encyclopedia of Polymer Science and Engineering , p 577. Wiley-Interscience,New Y ork (1988).20.D. J. V an Bramer, M. B. Shiflett, A.Y okozeki. U.S. Patent 5 345 013, 1994.21.D. A. Canelas and J. M. DeSimone. Adv. Polym. Sci. 133, 103–140 (1997).22.J. M. DeSimone, E. E. Maury, Y . Z. Menceloglu, J. B. McClain, T. R. Romack, J. R. Combes.Science 265, 356–359 (1994).23.Y .-L. Hsiao, E. E. Maury, J. M. DeSimone, S. M. Mawson, K. P . Johnston. Macromolecules 28,8159–8166 (1995).24.K. A. Shaffer, T. A. Jones, D. A. Canelas, J. M. DeSimone, S. P . Wilkinson. Macromolecules 29,2704–2706 (1996).25.D. A. Canelas, D.E. Betts, J. M. DeSimone. Macromolecules 29, 2818–2821 (1996).26.D. A. Canelas and J. M. DeSimone. Macromolecules 30, 5673–5682 (1997).27.D. A. Canelas, D.E. Betts, J. M. DeSimone, M. Z. Yates, K. P . Johnston. Macromolecules 31,6794–6805 (1998).28.H. Shiho and J. M. DeSimone. J. Polym. Sci. Part A: Polym. Chem. 37, 2429–2437 (1999).29.H. Shiho and J. M. DeSimone. J. Polym. Sci. Part A: Polym. Chem. 38, 1146–1153 (2000).30.T. Carson, J. Lizotte, J. M. DeSimone. Macromolecules 33, 1917–1920 (2000).31.H. S. Shiho and J. M. DeSimone. Macromolecules 33, 1565–1569 (2000).32.H. Shiho and J. M. DeSimone. J. Polym. Sci. Part A: Polym. Chem., submitted (2000).33.H.Shiho and J. M. DeSimone. Macromolecules , submitted (2000).34.M. Z. Yates, G. Li, J. J. Shim, S. Maniar, K. P . Johnston, K. T. Lim, S. Webber. Macromolecules 32, 1018–1026 (1999).35.A. I. Cooper, W. P . Hems, A.B. Holmes. Macromolecules 32, 2156–2166 (1999).36.A. I. Cooper and A.B. Holmes. Adv. Mater . 11, 1270 (1999).37.J. H. Xia, T. Johnson, S. G. Gaynor, K. Matyjaszewski, J. DeSimone. Macromolecules 32, 4802–4805 (1999).38.S. M. Gross, R. D. Givens, M. Jikei, J. R. Royer, S. Khan, J. M. DeSimone, P . G. Odell, G. K.Hamer. Macromolecules 31, 9090–9092 (1998).39.S. M. Gross, D. Flowers, G. Roberts, D. J. Kiserow, J. M. DeSimone. Macromolecules 32, 3167–3169 (1999).40.S. M. Gross, G. W. Roberts, D. J. Kiserow, J. M. DeSimone. Macromolecules 33, 40–45 (2000).陕西师范大学物理化学精品课程Green chemistry utilizing carbon dioxide for polymers1363© 2000 IUP AC, Pure and Applied Chemistry 72, 1357–136341.J. B. McClain, D. Londono, J. R. Combes, T. J. Romack, D. A. Canelas, D. E. Betts, G. D. Wignall,E. T. Samulski, J. M. DeSimone. J. Am. Chem. Soc. 118, 917–918 (1996).42. D. Chillura-Martino, R. Triolo, J. B. McClain, J. R. Combes, D. E. Betts, D. A. Canelas, E. T.Samulski, J. M. DeSimone, H. D. Cochran, J. D. Londono, G. D. Wignall J. Mol. Struct. 383, 3–10 (1996).43.Y . B. Melnichenko, E. Kiran, G. D. Wignall, K. D. Heath, S. Salaniwal, H. D. Cochran, M. Stamm.Macromolecules 32, 5344–5347 (1999).44.J. L. Fulton, D. M. Pfund, J. B. McClain, T. J. Romack, E. E. Maury, J. R. Combes, E. T. Samulski,J. M. DeSimone, M. Capel. Langmuir 11, 4241–4249 (1995).45.J. B. McClain, D. E. Betts, D. A. Canelas, E. T. Samulski, J. M. DeSimone, J. D. Londono, H. D.Cochran, G. D. Wignall, D. Chillura-Martino, R. Triolo. Science 274, 2049–2052 (1996).46.J. D. Londono, R. Dharmapurikar, H. D. Cochran, G. D. Wignall, J. B. McClain, D. E. Betts, D. A.Canelas, J. M. DeSimone, E. T. Samulski, D. Chillura-Martino, R. Triolo. J. Appl. Crystallogr . 30,690–695 (1997).47.E. Buhler, A. V . Dobrynin, J. M. DeSimone, M. Rubinstein. Macromolecules 31, 7347–7355(1998).48.M. L. O’Neill, M. Z. Yates, K. L. Harrison, K. P . Johnston, D. A. Canelas, D. E. Betts, J. M.DeSimone, S. P . Wilkinson. Macromolecules 30, 5050–5059 (1997).49.M. Z. Y ates, M. L. O’Neill, K. P . Johnston, S. Webber, D. A. Canelas, D. E. Betts, J. M. DeSimone.Macromolecules 30, 5060–5067 (1997).50.M. L. O’Neill, M. Z. Yates, K. P . Johnston, C. D. Smith, S. P. Wilkinson. Macromolecules 31,2838–2847 (1998).51.M. L. O’Neill, M. Z. Yates, K. P . Johnston, C. D. Smith, S. P. Wilkinson. Macromolecules 31,2848–2856 (1998).52.A. I. Cooper, J. D. Londono, G. Wignall, J.B. McClain, E. T. Samulski, J. S. Lin, A. Dobrynin, M.Rubinstein, A. L.C. Burke, J. M. J. Fréchet, J. M. DeSimone. Nature 389, 368 (1997).53.G. Stewart. In Green Chemistry Using Liquid and Supercritical Carbon Dioxide , J. M. DeSimone,W. Tumas, J. L. Y oung, and P. A. Anastas, (Eds.), Oxford University Press, Oxford, submitted for publication (2000).54.Y . Chernyak, F. Henon, E. Hoggan, B. Novick, J. DeSimone, R. Carbonell. In Green ChemistryUsing Liquid and Supercritical Carbon Dioxide , J. M. DeSimone, W. Tumas, J. L. Y oung, P . A.Anastas (Eds.), Oxford University Press, Oxford, submitted for publication (2000).陕西师范大学物理化学精品课程。