引物设计保护碱基列表
- 格式:doc
- 大小:82.50 KB
- 文档页数:4

11月13日引物合成的详解需要什么级其余引物?答:引物常用的纯化方式C18脱盐,OPC纯化,PAGE纯化,HPLC纯化。
依据实验需要,确立订购引物的纯度级别。
应用引物长度要求纯度级别要求一般PCR扩增<45base OPC一般PCR扩增>45base PAGE诊疗PCR扩增<40base OPC,PAGE DNA测序20base左右OPC亚克隆,点突变等依据实验要求定OPC,PAGE,HPLC基因建立(全基因合成)依据实验要求定PAGE反义核酸依据实验要求定PAGE修饰引物依据实验要求定PAGE,HPLC怎样计算引物的浓度?答:引物保留在高浓度的状况下比较稳固。
引物一般配制成10-50pmol/ul。
一般状况下,建议将引物的浓度配制成50pmol/ul,加水的体积(微升)按以下方式计算:V(微升)=OD数*(乘)33*(乘)(乘)20000/(除)引物的分子量。
引物的分子量能够从合成报告单上获取。
假如需要配制成其余浓度,按上述公式换算。
注意:1OD260=33ug/ml.怎样计算引物的Tm值?答:引物设计软件都能够给出Tm,与引物长度、碱基构成、引物使用缓冲的离子强度有关。
长度为25mer以下的引物,Tm计算公式为:Tm=4℃(G+C)+2℃(A+T)关于更长的寡聚核苷酸,Tm计算公式为:Tm=+xLog10[Na+]+(%GC)–600/size公式中,Size=引物长度。
怎样溶解引物?答:干燥后的引物质地特别松散,开盖前最好离心一下,或管垂直向上在桌面上敲敲,将引物粉末采集到管底。
依据计算出的体积加入去离子无菌水或10mMTris缓冲液,室温搁置几分钟,振荡助溶,离心将溶液采集到管底。
溶解引物用的水一般不要用蒸馏水,因为有些蒸馏水的pH值比较低(pH4-5),引物在这类条件下不稳固。
怎样保留引物?答:引物合成后,经过一系列办理和纯化步骤,旋转干燥而成片状物质。
引物在溶解前,室温状态下能够长久保留。
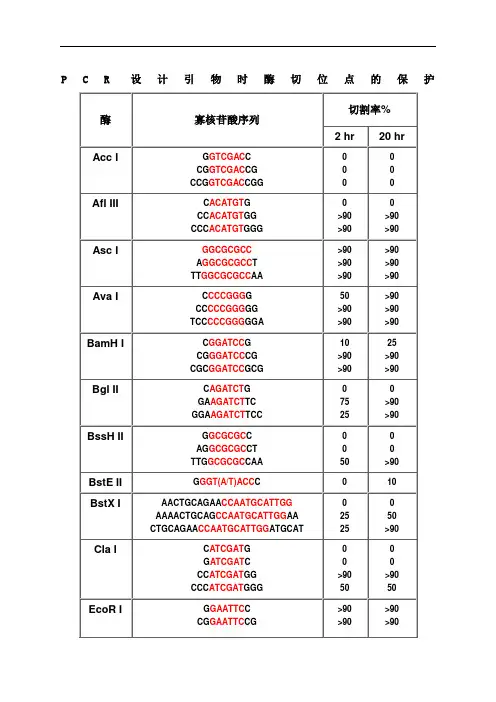
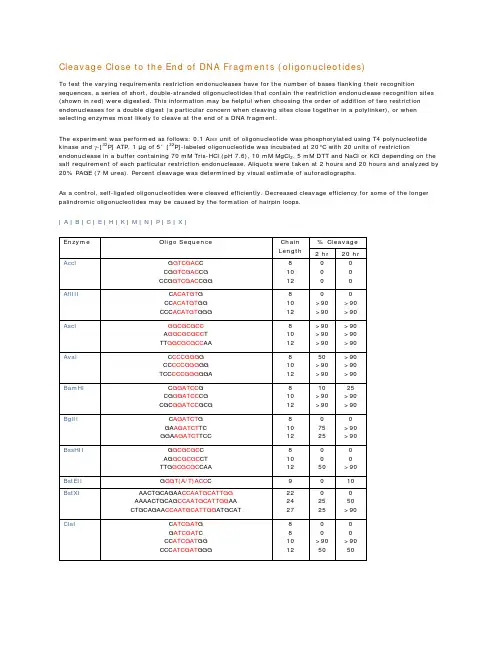
Cleavage Close to the End of DNA Fragments (oligonucleotides)To test the varying requirements restriction endonucleases have for the number of bases flanking their recognition sequences, a series of short, double-stranded oligonucleotides that contain the restriction endonuclease recognition sites (shown in red) were digested. This information may be helpful when choosing the order of addition of two restriction endonucleases for a double digest (a particular concern when cleaving sites close together in a polylinker), or when selecting enzymes most likely to cleave at the end of a DNA fragment.The experiment was performed as follows: 0.1 A260 unit of oligonucleotide was phosphorylated using T4 polynucleotide kinase and γ-[32P] ATP. 1 µg of 5´ [32P]-labeled oligonucleotide was incubated at 20°C with 20 units of restriction endonuclease in a buffer containing 70 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT and NaCl or KCl depending on the salt requirement of each particular restriction endonuclease. Aliquots were taken at 2 hours and 20 hours and analyzed by 20% PAGE (7 M urea). Percent cleavage was determined by visual estimate of autoradiographs.As a control, self-ligated oligonucleotides were cleaved efficiently. Decreased cleavage efficiency for some of the longer palindromic oligonucleotides may be caused by the formation of hairpin loops.| A | B | C | E | H | K | M | N | P | S | X |。

引物设计保护碱基列
表
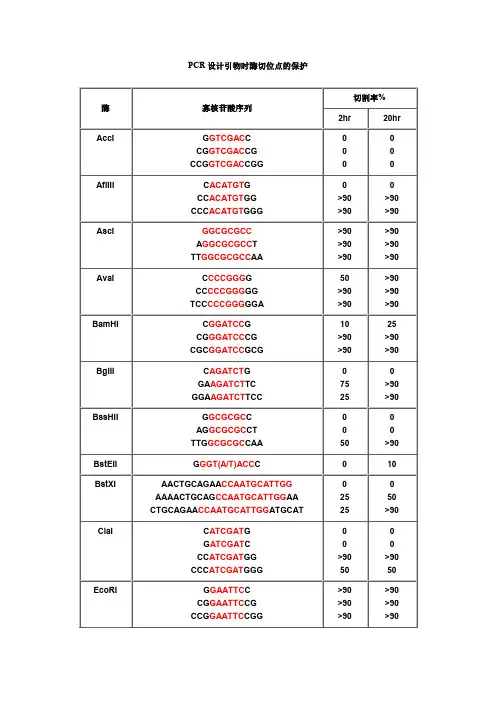
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的,见附表。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
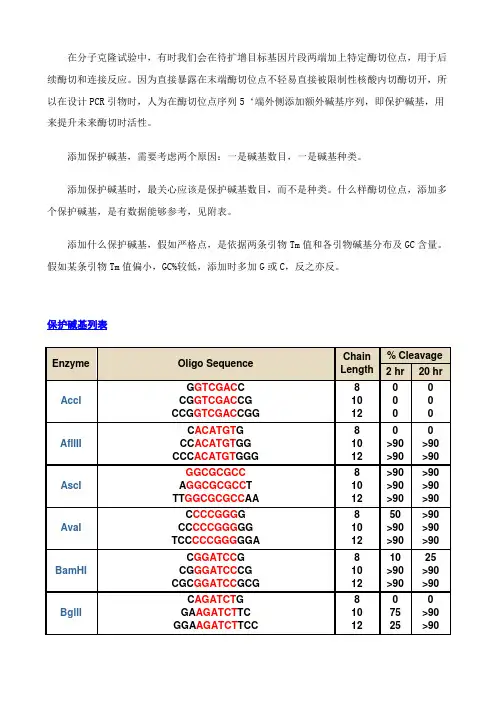
保护碱基列表。

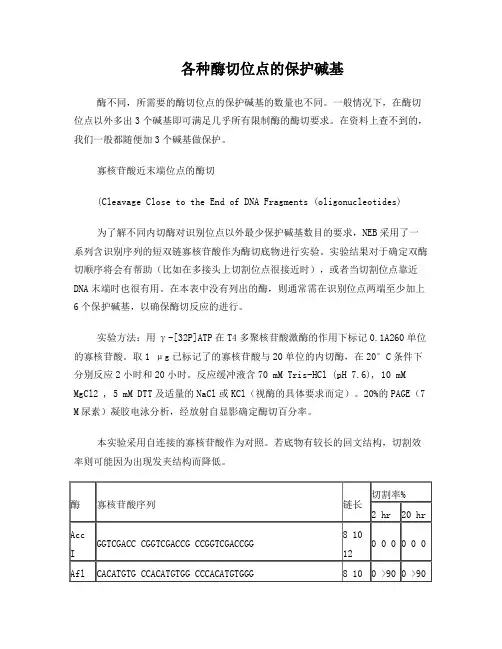
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments (oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同 buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。
在分子克隆试验中,有时我们会在待扩增目标基因片段两端加上特定酶切位点,用于后续酶切和连接反应。
因为直接暴露在末端酶切位点不轻易直接被限制性核酸内切酶切开,所以在设计PCR引物时,人为在酶切位点序列5‘端外侧添加额外碱基序列,即保护碱基,用来提升未来酶切时活性。
添加保护碱基,需要考虑两个原因:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心应该是保护碱基数目,而不是种类。
什么样酶切位点,添加多个保护碱基,是有数据能够参考,见附表。
添加什么保护碱基,假如严格点,是依据两条引物Tm值和各引物碱基分布及GC含量。
假如某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
保护碱基列表。

(完整)保护碱基列表
编辑整理:
尊敬的读者朋友们:
这里是精品文档编辑中心,本文档内容是由我和我的同事精心编辑整理后发布的,发布之前我们对文中内容进行仔细校对,但是难免会有疏漏的地方,但是任然希望((完整)保护碱基列表)的内容能够给您的工作和学习带来便利。
同时也真诚的希望收到您的建议和反馈,这将是我们进步的源泉,前进的动力。
本文可编辑可修改,如果觉得对您有帮助请收藏以便随时查阅,最后祝您生活愉快业绩进步,以下为(完整)保护碱基列表的全部内容。
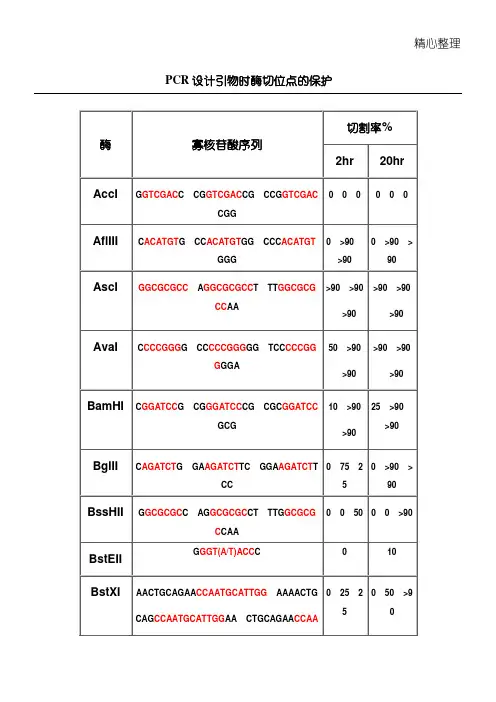
PCR设计引物时酶切位点的保护
注释:
1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
2.切割率:正确识别并酶切的效率
3.加保护碱基时最好选用切割率高时加的相应碱基。

酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在 酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上 查不到的,我们一般都随便加3个碱基做保护。
寡核昔酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides )为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB 采用了一系列含识别序列的 短双链寡核甘酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上 切割位点很接近时),或者当切割位点靠近DNA 末端时也很有用。
在本表中没有列出的酶,则通常 需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用Y -[32P]ATP 在T4多聚核甘酸激酶的作用下标记0.1A 260单位的寡核甘酸。
取1 pg 已标记了的寡核甘酸与20单位的内切酶,在20°C 条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCJ , 5 mM DTT 及适量的 NaCI 或 KCl (视酶的具体要求而定)。
20%的PAGE (7 M 尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核甘酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发 夹结构而降低。
切位点 的保护碱基2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶
切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,
即保护碱基,用来提高将来酶切时的活性。
添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的,见附表。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
保护碱基列表。