IVB和VB过渡族金属碳化物及其表面的结构和电子态的第一原理研究
- 格式:pdf
- 大小:925.25 KB
- 文档页数:7


CeCuGa3电子结构的第一性原理计算研究【摘要】我们采用基于密度泛函理论的第一性原理方法计算研究了CeCuGa3材料的电子结构。
我们计算确定了其基态磁结构,解释了其形成的原因。
【关键词】稀土金属Ce化合物磁结构费米面电子结构1 引言稀土金属Ce化合物由于具有重费米子行为,不同类型的磁有序等独特的物理性质而引起了科学研究的极大兴趣。
其中晶体结构为BaAl4的CeCuxGa4-x化合物最为代表。
最早报道CeCuGa3在3.5K温度下,其基态为铁磁态[1]。
另外Mentink 等人报道直到温度低到0.4K,CeCuGa3基态为顺磁态[2]。
而Martin等人通过对多晶CeCuGa3样品的研究,发现材料显示近藤晶格行为并且基态为反铁磁态[3]。
最近,Joshi等人再次通过实验对单晶CeCuGa3样品进行了晶体结构和磁学性质的研究,发现材料为4K以下的铁磁态[4]。
面对以上对于样品CeCuGa3相互矛盾的磁基态的报道,本文就采用基于密度泛函理论的vasp软件包对该材料的电子结构和磁学性质进行了计算并讨论了其磁基态性质。
2 模型构建和计算方法CeCuGa3晶体属于四方晶系结构,实验报道空间群为I4/mmm,No.139,如图1所示。
晶格常数a=b=4.273,c=10.44,α=β=γ=90°。
本文计算采用基于密度泛函理论(density functional theory,DFT)的V ASP(Vienna ab-initio simulation package)软件包进行计算。
计算步骤可以概括为三步:(1)对晶胞模型内部原子位置进行结构优化;(2)对材料进行磁构型计算,确定材料磁性基态。
(3)用广义梯度近似法(generalized gradient approximation,GGA)对优化后的理论模型进行单电子能量计算,对单电子能量计算结果进行总态密度(total density of states,TDOS)和分波态密度(partial density of states,PDOS)分析。
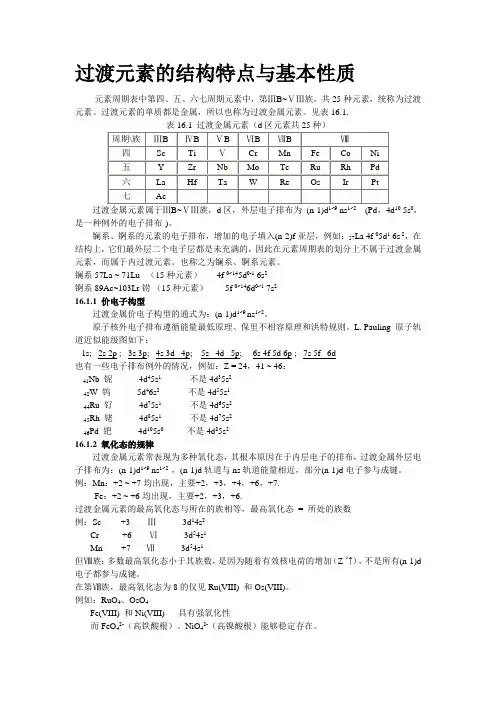
过渡元素的结构特点与基本性质元素周期表中第四、五、六七周期元素中,第ⅢB~ⅤⅢ族,共25种元素,统称为过渡元素。
过渡元素的单质都是金属,所以也称为过渡金属元素。
见表16.1.5s0,是一种例外的电子排布)。
镧系、锕系的元素的电子排布,增加的电子填入(n-2)f亚层,例如:57La 4f 05d1 6s 2,在结构上,它们最外层二个电子层都是未充满的,因此在元素周期表的划分上不属于过渡金属元素,而属于内过渡元素。
也称之为镧系、锕系元素。
镧系57La ~ 71Lu (15种元素) 4f 0~145d0-1 6s2锕系89Ac~103Lr铹(15种元素)5f 0~146d0~1 7s216.1.1 价电子构型过渡金属价电子构型的通式为:(n-1)d1~9 ns1~2。
原子核外电子排布遵循能量最低原理、保里不相容原理和洪特规则。
L. Pauling 原子轨道近似能级图如下:1s; 2s 2p ; 3s 3p; 4s 3d 4p; 5s 4d 5p; 6s 4f 5d 6p ; 7s 5f 6d也有一些电子排布例外的情况,例如:Z = 24,41 ~ 46:Nb 铌4d45s1不是4d35s241W 钨 5d46s2不是4d55s142Ru 钌4d75s1不是4d65s244Rh 铑4d85s1不是4d75s245Pd 钯4d105s0 不是4d85s24616.1.2 氧化态的规律过渡金属元素常表现为多种氧化态,其根本原因在于内层电子的排布,过渡金属外层电子排布为:(n-1)d1~9 ns1~2 ,(n-1)d轨道与ns轨道能量相近,部分(n-1)d电子参与成键。
例:Mn:+2 ~ +7均出现,主要+2,+3,+4,+6,+7.Fe:+2 ~ +6均出现,主要+2,+3,+6.过渡金属元素的最高氧化态与所在的族相等,最高氧化态= 所处的族数例:Sc +3 Ⅲ3d14s2Cr +6 Ⅵ3d54s1Mn +7 Ⅶ3d54s1但Ⅷ族:多数最高氧化态小于其族数,是因为随着有效核电荷的增加(Z *↑),不是所有(n-1)d 电子都参与成键。

Ag-La系二元化合物结构与电子性能的第一性原理研究高恩强;张照超;阮海光;黄福祥;陈志谦;王兰兰【摘要】通过第一性原理方法系统地研究了Ag-La系四种二元化合物的相结构稳定性和电子结构,包括B2-LaAg、LaAg2、La14Ag51和α-LaAg5。
结构优化后的平衡态晶体参数及质量密度与实验值相符。
结合能表明,随La浓度的增加,化合物的键合强度和稳定性提高。
Ag-La系4种二元化合物的生成焓分别为-21.7,-26.8,-22.9,-18.1kJ/mol,与实验值或CALPHAD的理论值相符。
电子结构表明这些化合物是导体,其原子间的价键性质是由金属键、离子键和共价键构成,其中:离子键是由Ag原子从La原子中得到电子形成的,共价键是由Ags-p及Agp-Lad杂化构成的。
并且这些化合物的共价键和离子键随着La浓度的增加而增强,使得Ag-La化合物稳定性提高。
【期刊名称】《重庆理工大学学报》【年(卷),期】2016(030)008【总页数】8页(P45-51,68)【关键词】Ag-La系稀土相电子结构第一性原理【作者】高恩强;张照超;阮海光;黄福祥;陈志谦;王兰兰【作者单位】[1]重庆理工大学材料科学与工程学院,重庆400054;[2]西南大学材料与能源学部,重庆400715【正文语种】中文【中图分类】O641由稀土与银构成的金属间化合物或者合金系化合物在工业应用和固态理论研究上受到广泛关注[1-10]。
在早期的研究中,Ag-La系二元化合物中存在LaAg、LaAg2和。
McMasters等[11]证实了LaAg3事实上是La14Ag51,并且存在LaAg5相,其中LaAg5具有两种存在形式:低温六方MgZn2类型结构和高温不确定的结构,但确定其结构同CeAg5、PrAg5及YbAg5。
低温相α-LaAg5与高温相β-LaAg5间的转变温度还未被确定,大概在770 K[13]。
LaAg2(CeCu2型)和La14Ag51(Gd14Ag51型)的晶体结构由文献[14-15]确定,F.Tambornino详细研究Gd14Ag51型化合物的晶体结构[7]。


大学无机化学第六章试题及答案第六章化学键理论本章总目标:1:掌握离子键、共价键和金属键的基本特征以及它们的区别;2:了解物质的性质与分子结构和键参数的关系;3:重点掌握路易斯理论、价电子对互斥理论、杂化轨道理论以及分子轨道理论。
4:熟悉几种分子间作用力。
各小节目标:第一节:离子键理论1:掌握离子键的形成、性质和强度,学会从离子的电荷、电子构型和半径三个方面案例讨论离子的特征。
2:了解离子晶体的特征及几种简单离子晶体的晶体结构,初步学习从离子的电荷、电子构象和半径三个方面来分析离子晶体的空间构型。
第二节:共价键理论1;掌握路易斯理论。
2:理解共价键的形成和本质。
掌握价键理论的三个基本要点和共价键的类型。
3:理解并掌握价层电子对互斥理论要点并学会用此理论来判断共价分子的结构,并会用杂化轨道理论和分子轨道理论来解释分子的构型。
第三节:金属键理论了解金属键的能带理论和三种常见的金属晶格。
第四节:分子间作用力1:了解分子极性的判断和分子间作用力(范德华力)以及氢键这种次级键的形成原因。
2;初步掌握离子极化作用及其强度影响因素以及此作用对化合物结构及性质的影响。
习题一选择题1.下列化合物含有极性共价键的是()(《无机化学例题与习题》吉大版)A.KClO3B.Na2O2C.Na2OD.KI2.下列分子或离子中键能最大的是()A.O2B.O2-C.O22+D.O22-3.下列化合物共价性最强的是()(《无机化学例题与习题》吉大版)A.LiIB.CIC.BeI2D.MgI24.极化能力最强的离子应具有的特性是()A.离子电荷高,离子半径大B.离子电荷高,离子半径小C.离子电荷低,离子半径小D.离子电荷低,离子半径大5.下列化合物中,键的极性最弱的是()(《无机化学例题与习题》吉大版)A.FeCl3B.AlCl3C.SiCl4D.PCl56.对下列各组稳定性大小判断正确的是()A.O2+>O22-B.O2->O2C.NO+>NOD.OF->OF7.下列化合物中,含有非极性共价键的离子化合物是()(《无机化学例题与习题》吉大版)A.H2O2B.NaCO3C.Na2O2D.KO38.下列各对物质中,是等电子体的为()A.O22-和O3B.C和B+C.He和LiD.N2和CO9.中心原子采取p2杂化的分子是()(《无机化学例题与习题》吉大版)A.NH3B.BCl3C.PCl3D.H2O10.下列分子中含有两个不同键长的是()A.CO2B.SO3C.SF4D.某eF411.下列分子或离子中,不含有孤电子对的是()(《无机化学例题与习题》吉大版)A.H2OB.H3O+C.NH3D.NH4+12.氨比甲烷易溶于水,其原因是()A.相对分子质量的差别B.密度的差别C.氢键D.熔点的差别13.下列分子属于极性分子的是()(《无机化学例题与习题》吉大版)l4B.CH3OCH3C.BCl3D.PCl514.下列哪一种物质只需克服色散力就能使之沸腾()A.HClB.CH3Cll4D.NH315.下列分子中,中心原子采取等性杂化的是()(《无机化学例题与习题》吉大版)A.NCl3B.SF4C.CHCl3D.H2O16.下列哪一种物质既有离子键又有共价键()2A.NaOHB.H2OC.CH3ClD.SiO217.下列离子中,中心原子采取不等性杂化的是()(《无机化学例题与习题》吉大版)A.H3O+B.NH4+C.PCl6-D.BI4-18.下列哪一种分子的偶极矩最大()A.HFB.HClC.HBrD.HI19.下列分子中,属于非极性分子的是()(《无机化学例题与习题》吉大版)A.SO2B.CO2C.NO2D.ClO220.下列分子或离子中,中心原子的杂化轨道与NH3分子的中心原子轨道最相似的是()(《无机化学例题与习题》吉大版)A.H2OB.H3O+C.NH4+D.BCl321.下列分子或离子中,构型不为直线形的是()(《无机化学例题与习题》吉大版)A.I3+B.I3-C.CS2D.BeCl222.下列分子不存在Ⅱ键的是()(《无机化学例题与习题》吉大版)A.COCl2B.O3C.SOCl2D.SO323.下列分子中含有不同长度共价键的是()(《无机化学例题与习题》吉大版)A.NH3B.SO3C.KI3D.SF424.下列化合物肯定不存在的是()(《无机化学例题与习题》吉大版)A.BNB.N2H4C.C2H5OHD.HCHO二填空题1.比较大小(《无机化学例题与习题》吉大版)(1)晶格能AlF3AlCl3NaClKCl(2)溶解度CuF2CuCl2Ca(HCO3)NaHCO32.NO+、NO2、NO2-的几何构型分别是、、、其中键角最小的是3.给出晶宝包中离子总数:立方ZnS;NaCl;CCl(《无机化学例题与习题》吉大版)4.CO2是分子;SO2是分子;BF3是分子;NF3是分子;PF5是分子。

物理所硕士招生专业及研究方向理论物理主要研究方向1、高温超导体机理、BEC理论及自旋电子学相关理论研究。
2、凝聚态理论;3、原子分子物理、量子光学和量子信息理论;4、统计物理和数学物理。
5、凝聚态物理理论、计算材料、纳米物理理论6、自旋电子学,Kondo效应。
7、凝聚态理论、第一原理计算、材料物性的大规模量子模拟。
8、玻色-爱因斯坦凝聚, 分子磁体, 表面物理,量子混沌。
凝聚态物理主要研究方向1、非常规超导电性机理,混合态特性和磁通动力学。
(1)高温超导体输运性质,超导对称性和基态特性研究。
(2)超导体单电子隧道谱和Andreev反射研究。
(3)新型Mott绝缘体金属-绝缘基态相变和可能超导电性探索。
(4)超导体磁通动力学和涡旋态相图研究。
(5)新型超导体的合成方法、晶体结构和超导电性研究。
2、高温超导体电子态和异质结物理性质研究(1)高温超导体和相关氧化物功能材料薄膜和异质结的生长的研究。
(2)铁电体极化场对高温超导体输运性质和超导电性的影响的研究。
(3)高温超导体和超大磁电阻材料异质结界面自旋极化电子隧道效应的研究。
(4)强关联电子体系远红外物性的研究。
3、新型超导材料和机制探索(1)铜氧化合物超导机理的实验研究(2)探索电子―激子相互作用超导体的可能性(3)高温超导单晶的红外浮区法制备与物理性质研究4、氧化物超导和新型功能薄膜的物理及应用研究(1)超导/介电异质薄膜的制备及物性应用研究(2)超导及氧化物薄膜生长和实时RHEED观察(3)超导量子器件的研究和应用(4)用于超导微波器件的大面积超导薄膜的研制5、超导体微波电动力学性质,超导微波器件及应用。
6、原子尺度上表面纳米结构的形成机理及其输运性质(1)表面生长的动力学理论;(2)表面吸附小系统(生物分子,水和金属团簇)原子和电子结构的第一性原理计算;(3)低维体系的电子结构和量子输运特性(如自旋调控、新型量子尺寸效应等)。
.7、III-V族化合物半导体材料及其低维量子结构制备和新型器件探索(1)宽禁带化合物(In/Ga/AlN,ZnMgO)半导体及其低维量子结构生长、物性、微结构以及相互关系的研究,宽禁带化合物半导体新型微电子、光电子器件探索;(2)砷化镓基、磷化铟基新型低维异质结材料的设计、生长、物性研究及其新型微电子/光电子器件探索;(3)SiGe/Si应变层异质结材料的制备及物性研究。

二硒化钼合成与应用的研究进展亓淑艳;刘小虎;陈明【摘要】过渡族金属二硫化物由于其特殊的结构和性能,已成为国内外研究的重点.二硒化钼性能优异,应用广泛,是过度族金属二硫化物中极具代表性的物质之一.综述了二硒化钼的结构、性质,从固相法、气相法、液相法三方面入手,评述了二硒化钼的制备现状,并对各种制备方法的优缺点进行了比较.另外对二硒化钼在材料改性、润滑剂添加剂和场效应晶体管等方面的应用作了详细的介绍,并展望了二硒化钼在光催化剂和太阳能电池领域的应用和发展.【期刊名称】《哈尔滨理工大学学报》【年(卷),期】2016(021)001【总页数】6页(P22-26,30)【关键词】二硒化钼;合成;应用【作者】亓淑艳;刘小虎;陈明【作者单位】哈尔滨理工大学材料科学与工程学院,黑龙江哈尔滨150080;哈尔滨理工大学材料科学与工程学院,黑龙江哈尔滨150080;哈尔滨理工大学材料科学与工程学院,黑龙江哈尔滨150080【正文语种】中文【中图分类】O612.6过渡族金属二硫化物是由大量的二维层状金属二硫化物构成的,是由 IVB-VB-VIB-VIA元素形成的化合物.在许多领域比如光催化、能量储存、固体润滑、微电子和光电领域有着广泛的应用[1-6].另外,与石墨烯比较过渡族金属二硫化物的禁带宽度略宽,在场效应晶体管和低功率电子等方面发挥较为突出的作用.单层的过渡族金属二硫化物有很强的自旋轨道耦合和反演对称的破坏性,这也完全符合自旋电子学在应用方面对半导体要求[7-11].二硒化钼就是过渡族金属二硫化物中的一种.二硒化钼不仅拥有上述过渡族金属二硫化物的所有特性,另外与其他二硫化物相比,二硒化钼还拥有背栅效应、固体润滑等性能.二硒化钼是灰黑色的共价化合物,属于六方晶系,具有类似于三明治的片层状结构,层与层之间通过微弱的范德华力松散得结合在一起,相邻的俩分子层可以相互滑动,具有较低的摩擦系数,常用作固体润滑剂,具有降低材料的磨损,减少机械设备运转阻力,降低能耗,延长使用寿命的功能;另外其化学性质稳定,可用于高温高压润滑剂[12-13].同时,MoSe2中每个钼原子被六个硒原子包围,呈三角棱柱状,Mo-Se 棱面相当多,比表面积大,表面活性高,具有优异的催化活性,广泛用作石油加工行业的加氢脱硫催化剂,以及废水处理催化剂[ 14-16].由于具有较大层间距,一些离子如Li+和Na+可以很容易地插入到二硒化钼的层间,形成插层化合物,在材料改性、制备新型功能材料方向具有极大的应用和发展.近几年,人们发现当将二硒化钼的厚度限定为单分子层时,间接禁带宽度的半导体可以转化为直接禁带宽度的半导体,光致发光的强度明显提高,如此使得其拥有在光电领域应用的极大潜能,比如光电探测器[17-19].二硒化钼在诸多方面表现出的优异性能引起了国内外科学家极大兴趣.近年来研究人员采用不同方法制备出了各种形貌的二硒化钼,有人甚至制出单分子层的二硒化钼.合成二硒化钼的方法很多,按制备状态分为固相法、气相法、液相法;另外按反应机理还分为还原法、分解法、氧化法、电化学法;按反应方式的不同分为物理法、化学法及物化综合法.本文主要从固相法、气相法、液相法方面对二硒化钼的制备方法进行介绍.2.1 固相法固相法是用合适的原料,在固态下通过物理或化学的方法制得二硒化钼的方法.该方法的突出特点是操作方便,合成工艺简单,转化率高,粒径均匀,污染少,可避免或减少液相中易出现的硬团聚现象,以及由中间步骤和高温反应引起的粒子团聚现象.宋也黎等[20]用纯度 99.9% 的钼粉和99.9% 的硒粉,将两者按照化学计量比(1∶3)混合,在特制的反应釜中750℃下反应,制备出长为100~500 nm, 厚为10~50 nm的片状的MoSe2纳米粒子.2.2 气相法2.2.1 化学气相沉积法化学气相沉积法是传统的制备薄膜的技术,其原理是利用气态的先驱反应物,通过原子、分子间化学反应,使得气态前驱体中的某些成分分解,而在基体上形成薄膜.化学气相沉积包括常压化学气相沉积、等离子体辅助化学沉积、激光辅助化学沉积、金属有机化合物沉积等.在制备单分子层产物时化学气相沉积法具有其独特性.到目前为止,在众多制备单分子层状过渡族金属二硫化物的方法中,化学气相沉积法是最为优异的,因为该方法不仅能在多种基底如铜箔、青玉、云母、硅等基底上大面积制备需要的单分子层产物,且对产物的特性不会造成影响.而其他方法如流体剥离法和锂基化学剥离法都需要用到有机溶液,这样可能会影响到样品的品质.另外通过激光,等离子体,或退火处理也可以得到单层薄片,但这种减薄过程也可能会一定程度地损坏样品表面或降低样品的结晶度.Xin Lu,M. 等[21]通过对MoO3的硒化在SiO2/Si基底上制备出了厚度分别为0.8 nm和1.5 nm单分子层和双分子层的MoSe2.Jonathan C.等[22]用Se和MoO3 作为化学气相供给,在300 nm的衬底上制备出了光学可区别的、高度结晶的、单层和多层的MoSe2纳米片,尤其是在边长为30 μm的三角形区域它可以进一步发展合并成连续薄膜.在该反应中H2发挥了极其重要的作用,他不仅作为载气使得MoSe2在SiO2/Si基底上沉积,而且还是该反应所需要的还原剂,将正六价的钼还原到正四价.单层的MoSe2在1.55ev处存在一个很强的近边带发射,而双分子层和多分子层的MoSe2的边带发射却非常微弱.这表明过渡到直接带隙的半导体的厚度变为单层.2.2.2 化学气相传输法该方法是通过控制源区和生长端的温度梯度, 使用碳辅助增强质量传输效应,在无籽晶自发成核的条件下,得到目标晶体.Moussa Bougoumaa等[23]将硒粉和钼粉放置在石英玻璃管中,控制温度梯度为980-1020℃,以TeCl4为载气来合成MoSe2.源区的硒粉和钼粉在高温下气化并反应生成MoSe2,在生长端,MoSe2形成单晶和多晶的粉末.气相传输法具有操作简单、生长成本低等优点,但对设备的要求较高.2.3 液相法2.3.1 低温液相沉积法低温液相沉积法制造固体催化剂的方法之一,即将金属盐水溶液和沉淀剂分别加入搅拌罐中,生成固体沉淀.该工艺主要包括沉淀的生成和固液分离,其中沉淀的生成是该工艺的关键步骤.这个方法简单易行,但纯度低,颗粒半径大.P.P. Hankare 等[24]在低温下(冰水混浴中)将钼酸铵、硒代硫酸钠、水合肼、柠檬酸放置于碱性溶液中,以50 rap/min的速度搅拌9 h,在圆底烧瓶的底部生成了灰黑色的沉淀即MoSe2,其禁带宽度大约为1.43 ev,具有n型传导机制,电导率为10-2(Ωcm)-1.另外,该实验中用到的试剂硒代硫酸钠化学性质活泼,极易被空气中的氧气氧化,在制备好后应立即使用或密封避光保存.2.3.2 抑制性沉淀法与其他反应方法相比,抑制性沉淀技术具有很大的优势.首先其制备成本较低,不需要昂贵反应试剂和设备,且工艺流程短操作简单.其次该反应在常温常压下即可完成,不需要高温高压等苛刻的条件;而且利用该方法可以大量制备所需要的产物.最后该方法可以定性和定量的控制反应沉积物,达到了人为设计目标产物的目的.S. N. Gawale等[25]用该方法将MoSe2薄片沉积在玻璃或是不锈钢的基底上.该反应是把连二硫酸钠、MoO3、三乙胺、硒代硫酸钠置于碱性溶液中在333K的温度下反应,得到晶粒分布均匀、紧凑且没有销孔,禁带宽度为1.76eV ,的六方晶体结构MoSe2.其电导率的温度依赖性因不同的导电区域而异且在表面产生的热电压是几个微伏级表明它为n型传导机制.2.3.3 其他液相法Brian C. Helmly等[26]以Mo(CO)6为钼源、硒粉为硒源在二甲苯溶剂中反应合成二硒化钼,整个反应在氩气气氛中进行.该反应制备出纯度很高的MoSe2,通过紫外-可见光谱分析可知产物的粒径大约在10-30 nm之间.我们知道金属钼的熔沸点和硬度很高,反应活性非常低;非金属硒是氧族元素,但是其分子结合力很弱,在参与反应时无法与硫和氧的氧化能力相媲美.所以直接将Se和Mo化合合成MoSe2需要极高的温度和压力才能达到.可是氧化Mo(CO)6等低价配合物克服了上述缺点,而且能够生产粒径较小的粉末或是沉积成均匀的薄片.这是由于配合物Mo(CO)6中有σ配位键与π反馈键,这样就使得钼原子的外层的电子结构发生了极大的变化,更容易失去电子,反应活性更高,在合成目标产物时并不需要十分苛刻的条件.同时Mo(CO)6等配合物的熔沸点不高,很容易脱离原来含有杂质的体系,所以容易获得较高纯度的产物.另外,Mo(CO)6熔点150℃左右,所以它既可以在液态中反应也可以在气态中反应.当然该方法也有其不足之处,那就是Mo(CO)6具有极高的毒性,吸入、皮肤接触及吞食都会造成极大的危害严重的可能会导致生命危险.另外,该反应生成的有毒性气体CO容易与人体的血红蛋白结合使人中毒,所以该反应必须在通风良好的情况下操作[27].3.1 材料改性方面的应用3.1.1 制备插层化合物由于MoSe2具有类似于石墨的层状结构,层与层间通过范德华力作用,作用力较弱,在一定条件下,一些异类原子或分子或无机、有机的离子可以通过吸附、插入、夹入、悬挂、柱撑、嵌入等方式破坏分子间力进入MoSe2层状化合物的层间而不破坏其层状结构,这样化合形成一种新型插层材料具有原材料所没有的特异性能,这些功能材料能够弥补传统材料在新形势下的应用缺陷.Hao Xu等 [28]将MoSe2和正丁基锂在手套式操作箱中搅拌12天,形成了LixMoSe2插层化合物.之后作者用合成的LixMoSe2分别与聚苯胺、聚丙基烯酸甲酯、苯基乙胺、苯丙醇胺在超声波作用下,长时间反应,最后形成了对应聚合物-MoSe2插层化合物,与主体化合物MoSe2相比,在室温下他们的电导率都有显著提高.3.1.2 化合物的掺杂半导体的常用掺杂技术主要有两种,即高温(热)扩散和离子注入.掺入的杂质主要有两类:第一类是提供载流子的受主杂质或施主杂质(如B、P、Re);第二类是产生复合中心的重金属杂质(如Au、Ag、Pt).热扩散技术需较高温度,通常是在几百甚至上千摄氏度的温度下进行.离子注入技术掺杂时,即使在较低温度下也能够使重金属杂质很容易地通过晶格间隙而进入到半导体中去,所以扩散的温度一般较低.S.Y. Hu等[29]以化学气相传输法(属于高温扩散法)将硒粉、钼粉、Re置于石英管的源区,控制温度梯度为930-960℃,生成的MoSe2晶体原子在高温下的热运动加剧,以使得MoSe2晶体中的原子获得足够高的能量而离开晶格位置、留下空位,Re原子的热运动同样剧烈,这就有利于Re进入MoSe2晶体内部形成掺杂化合物.通过测试,掺杂Re的MoSe2晶体直接和间接带隙红移,晶体及其电导率的各项异性大幅度地降低.3.1.3 化合物负载在催化剂的光催化体系中引入贵金属后,贵金属作为光生电子的接收器,可促进复合系统界面载流子的运输,使光生电子在金属表面积累,而空穴则留在催化剂表面,降低了光生电子-空穴的复合率,提高了催化剂的光催化活性.还能够提升催化剂利用率.通过催化剂的负载,可以使催化剂分子充分的分散开来这样就能增大催化剂的比表面积,从而催化剂活性种的数目也就会增加.而且催化剂的负载化,使得催化剂的初始活性有一定的降低,而一定程度上可以解决在聚合过程中的因初始聚合过快而产生的暴聚问题.并且,负载化的催化剂的活性中心一般很稳定,能够长时间的保持催化活性,因而也能够提高聚合物的产量.负载贵金属可以采用普通浸渍还原法,即将半导体薄膜或颗粒浸渍在含有贵金属盐的溶液中,然后用还原剂高温还原;另外,还可以采用光还原法,即将半导体薄膜浸渍在贵金属盐和牺牲有机物如甲醇、乙酸等溶液中,然后用紫外光照射将贵金属还原而沉积在半导体表面.Su S等[30]采用浸渍法将Au, Ag 和Pt 负载到MoSe2表面上,通过测试发现Au等贵金属均匀的负载在MoSe2表面,化合物中的金属离子显示了其特有的表面等离激元带.该复合材料在光催化等领域都有着广泛的应用.3.2 固体润滑剂方面的应用MoSe2具有类似于石墨的晶体结构,层与层之间是通过范德华力作用的,层间容易滑动,形成平行于摩擦表面排列的组织结构,有利于自润滑功能.Mo原子和Se 原子之间以离子键结合,使MoSe2润滑膜的强度较高,能够防止润滑剂在金属的突出部位破损.Se原子暴露在MoSe2晶体表面,对金属表面产生很强的黏附作用.纳米MoSe2微球具有优异的自润滑特性和超低摩擦特性.宋也黎等[20]通过在相同的基础润滑油中添加MoSe2和MoS2发现添加MoSe2纳米片的基础油的摩擦因数要比添加MoS2纳米基础油的高, 其中 MoSe2 添加 5% ( 质量分数) 的润滑油的摩擦性能最好.MoSe2可以降低磨损,改善材料的摩擦性能.3.3 场效应晶体管方面的应用通过改变外加垂直于半导体表面上电场的方向或大小,以控制半导体导电层(沟道)中多数载流子的密度或类型.它是由电压调制沟道中的电流,其工作电流是由半导体中的多数载流子输运.这类只有一种极性载流子参加导电的晶体管又称单极型晶体管.与双极型晶体管相比,场效应晶体管具有输入阻抗高、噪声小、极限频率高、功耗小,制造工艺简单、温度特性好等特点,广泛应用于各种放大电路、数字电路和微波电路等.Babak Fallahazad等[31]用机械剥离法制备出超薄的MoSe2进而制备出了背栅场效应晶体管.MoSe2场效应晶体管是n型半导体且具备高栅极调制,拥有极大的开关比(超过了106).该晶体管在源极和漏极交换方面显示出非对称性的特征,这个现象可以用金属和MoSe2接触面的肖特基势垒来解释.在众多制备MoSe2的方法中,固相法、液相法或气相法都各有优缺点.固相法合成工艺简单,转化率高,粒径均匀,污染少,但是反应条件苛刻,对设备的要求较高.液相法不需要较高的温度和压力,有的方法甚至在常温常压下就可以实施,可操作性强.但是在制备过程中很容易出现硬团聚现象,且制备工艺流程长.气相法工艺简单,反应迅速,但对设备要求较高,且产品纯度不高.因此实验方案的制定和实验方法的选取,要根据制备产物的粒径、纯度等要求和现实的实验条件来确定.应用方面,MoSe2在制备各种材料和场效应晶体管、用作润滑剂添加剂等方面较为成熟.其他方面还有待进一步研究.但是可以预见,在未来发展中MoSe2在光催化和太阳能电池等领域的应用将拥有极大的发展.与现在最常见的光催化材料TiO2相比MoSe2的禁带宽度更窄,大约为1.4ev[32],能够吸收可见光区和近红外光驱的光,这是二硒化钼成为广泛应用的光催化材料的一个前提.另外,MoSe2有很高的电导率,电导率大约为50(Ωcm)-1[33]可以用来做太阳能电池的电极.目前,对二硒化钼的制备和应用的研究还仅仅处于初始阶段,离大面积地推广应用还有一段距离,需要国内外科研工作者继续努力.【相关文献】[1] RAO B G, MATTE H S S R, RAO C N R. Decoration of Few-Layer Graphene-Like MoS2 and MoSe2 by Noble Metal Nanoparticles[J]. Journal of Cluster Science, 2012, 23(3): 929-937.[2] MARGULIS L, SALITRA G, TENNE R, et al. Nested Fullerene-like Structures[J]. Nature, 1993, 365(6442): 113-114.[3] MONDAL B, SENGUPTA K, RANA A, et al. Cobalt Corrole Catalyst for Efficient Hydrogen Evolution Reaction from H2O under Ambient Conditions: Reactivity, Spectroscopy, and Density Functional Theory Calculations[J]. Inorganic chemistry, 2013, 52(6): 3381-3387.[4] KIM Y, HUANG J L, LIEBER C M. Characterization of Nanometer Scale Wear and Oxidation of Transition Metal Dichalcogenide Lubricants by Atomic Force Microscopy[J]. Applied physics letters, 1991, 59(26): 3404-3406.[5] LI Y, WANG H, XIE L, et al. MoS2 Nanoparticles Grown on Graphene: an Advanced Catalyst for the Hydrogen Evolution Reaction[J]. Journal of the American Chemical Society, 2011, 133(19): 7296-7299.[6] HUANG X, ZENG Z, ZHANG H. Metal Dichalcogenide Nanosheets: Preparation, Properties and Applications[J]. Chemical Society Reviews, 2013, 42(5): 1934-1946.[7] XIAO D, LIU G B, FENG W, et al. Coupled Spin and Valley Physics in Monolayers of MoS2 and other group-VI dichalcogenides[J]. Physical Review Letters, 2012, 108(19): 196802.[8] HE K, POOLE C, MAK K F, et al. Experimental Demonstration of Continuous Electronic Structure Tuning Via Strain in Atomically Thin MoS2[J]. Nano letters, 2013, 13(6): 2931-2936.[9] XIN X, WEIDE Z. Photocatalysis of Carbon Nanotubes/Semiconductor Composites[J]. Progress in Chemistry, 2011, 23(4): 657-668.[10]ZHANG J, XU J X, KONG Y Z, et al. Generation of Chemical-inducible Activation Tagging T-DNA Insertion Lines of Arabidopsis Thaliana[J]. Acta Genetica Sinica, 2005,32(10): 1082-1088.[11]EVANS B L, HAZELWOOD R A. Optical and Structural Properties of MoSe2[J]. Physica Status Solidi, 1971, 4(1):181-192.[12]HARPENESS R, GEDANKEN A, Weiss A M, et al. Microwave-assisted Synthesis of Nanosized MoSe2[J]. Journal of Materials Chemistry, 2003, 13(10):2603-2606.[13]SHI Y, HUA C, LI B, et al. Highly Ordered Mesoporous Crystalline MoSe2 Material with Efficient Visible-Light-Driven Photocatalytic Activity and Enhanced Lithium Storage Performance[J]. Advanced Functional Materials, 2013, 23(14):1832-1838.[14]MEINHOLD H, WEISER G. Modulation Spectroscopy on MoS2, and MoSe2[J]. Physica Status Solidi B Basic Research, 2006, 73(1):105-115.[15]HANKARE P P, PATIL A A, CHATE P A, et al. Characterization of MoSe2 Thin Film Deposited at Room Temperature From Solution Phase[J]. Journal of Crystal Growth, 2008, 311(1):15-19.[16]KRISHNAN R, PAYZANT E A, KACNYZKI R, et al. Reaction Kinetics and Pathways of MoSe2[J]. Conference Record of the IEEE Photovoltaic Specialists Conference, 2010,12(1):001006-001008.[17]KUC A, ZIBOUCHE N, HEINE T. Influence of Quantum Confinement on the Electronic Structure of the Transition Metal Sulfide TS 2[J]. Physical Review B, 2011, 83(24): 245213.[18]RAO C N R, SUBRAHMANYAM K S, MATTE H S S R, et al. A Study of the Synthetic Methods and Properties of Graphenes[J]. Science and Technology of Advanced Materials, 2010, 11(5): 054502..[19]LIU L, BALA Kumar S, OUYANG Y, et al. Performance Limits of Monolayer Transition Metal Dichalcogenide Transistors[J]. Electron Devices, IEEE Transactions on, 2011, 58(9): 3042-3047.[20]宋也黎,彭红红,李长生,等. MoSe2纳米片的制备及其摩擦性能[J]. 机械工程材料,2011,1:83-85.[21]KONG D, WANG H, CHA J J, et al. Synthesis of MoS2 and MoSe2 Films with Vertically Aligned Layers[J]. Nano Letters, 2013, 13(3): 1341-1347.[22]SHAW J C, ZHOU H, CHEN Y, et al. Chemical Vapor Deposition Growth of Monolayer MoSe2 Nanosheets[J]. Nano Research, 2014, 7(4): 1-7.[23]BOUGOUMA M, BATAN A, GUEL B, et al. Growth and Characterization of Large, High Quality MoSe2 Single Crystals[J]. Journal of crystal growth, 2013, 363: 122-127.[24]HANKARE P P, PATIL A A, CHATE P A, et al. Characterization of MoSe 2 Thin Film Deposited at Room Temperature from Solution Phase[J]. Journal of Crystal Growth, 2008, 311(1): 15-19.[25]WANG Q H, KALANTAR-ZADEH K, KIS A, et al. Electronics and Optoelectronics of Two-dimensional Transition Metal Dichalcogenides[J]. Nature Nanotechnology, 2012,7(11): 699-712.[26]HELMLY B C, LYNCH W E, NIVENS D A. Preparation and Spectroscopic Characterization of MoS2 and MoSe2 Nanoparticles[J]. Spectroscopy Letters, 2007, 40(3): 483-492.[27]胡坤宏, 沃恒洲, 韩效钊, 等. 纳米二硫化钼制备现状与发展趋势[J]. 现代化工, 2003, 23(8): 14-17.[28]XU H, BISSESSUR R, DAHN D C. Nanomaterials Based on Polyanilines and MoSe2[J]. Journal of Inorganic and Organometallic Polymers and Materials, 2014, 24(1): 219-225. [29]HU S Y, QIU J, ZHANG C H, et al. The Effect of Chaihu-Shugan-San and Its Components on the Expression of ERK5 in the Hippocampus of Depressed Rats[J]. Journal of Ethnopharmacology, 2014.[30]SU S, SUN H, XU F, et al. Direct Electrochemistry of Glucose Oxidase and a Biosensor for Glucose Based on a Glass Carbon Electrode Modified with MoS2 Nanosheets Decorated with Gold Nanoparticles[J]. Microchimica Acta, 2014: 1-7.[31]LARENTIS S, TOLSMA J R, FALLAHAZAD B, et al. Band Offset and Negative Compressibility in Graphene-MoS2 Heterostructures[J]. Nano letters, 2014, 14(4): 2039-2045.[32]HANKARE P P, PATIL A A, CHATE P A, et al. Characterization of MoSe 2 Thin Film Deposited at Room Temperature from Solution Phase[J]. Journal of Crystal Growth, 2008, 311(1): 15-19.[33]SATHED D.J., HANKARE P.P., MANIKSHETE A.H., et al. Patil Structural, Optical and Microscopic Studies of Tungsten Substituted Molybdenum Diselenide Thin Films[J]. Journal of Alloys and Compounds, 2010, 499(2):187-193.。

可变单元c-neb方法-回复什么是可变单元cneb方法?可变单元cneb方法(Constrained Natural Extensional Basis, CNEB)是一种计算化学方法,用于研究分子和化学反应的势能面。
在化学反应中,分子会从一个能量极小值的构象转变为另一个能量极小值构象,这种转变称为化学反应路径。
CNEB方法可以用于确定最小能量路径,同时还能够研究该路径上的过渡态以及反应的动力学性质。
CNEB方法的基本原理是通过过渡态理论和多体势能外推法来构建势能面。
过渡态理论指的是研究化学反应路径中过渡态(transition state)的理论,通过确定过渡态的几何结构和能量来描绘反应的进程。
而多体势能外推法是一种根据已知构象和势能计算其他构象的方法。
CNEB方法将这两种方法结合起来,先通过过渡态理论确定反应路径上的过渡态,并通过势能表面外推计算路径上的其他构象。
CNEB方法的关键步骤包括以下几个:1. 选择初始构象:首先需要选择一个初始构象作为化学反应路径的起点。
这通常可以通过分子力学或量子化学计算得到。
2. 猜测中间构象:根据初始构象和目标构象,通过给定的方法猜测中间构象。
这些中间构象作为过渡态的候选构象。
3. 计算过渡态的几何结构:对于每个中间构象,使用过渡态理论计算其几何结构和能量。
常见的过渡态搜索方法有采用内禀反应坐标(IRC)法、能量梯度法等。
4. 多体势能外推:通过已知构象和势能表面计算路径上其他构象的势能。
这需要根据分子力学或量子化学方法计算每个构象的势能。
5. 更新构象:根据计算得到的势能,选择能量最低的构象作为路径上的一个点,并作为下一步计算的初始构象。
然后重复步骤2-5,直到达到收敛条件。
6. 确定最小能量路径:通过计算路径上每个构象的能量,可以确定最小能量路径。
这个路径给出了化学反应的过渡态和构象变化情况。
CNEB方法的优点在于可以通过对构象进行优化和过渡态搜索,找到势能表面上最小能量的化学反应路径。

“第一性原理研究”资料合集目录一、金属间化合物相稳定性、层错能及力学性质的第一性原理研究二、基于二维材料的高效锂硫电池催化剂的第一性原理研究三、第一性原理研究CH3NH3SnCl3钙钛矿太阳能电池的光伏特性和载流子迁移率四、过渡金属氧化物电子结构与性质的第一性原理研究五、碱金属、碱土金属以及金属钍的硼碳化物第一性原理研究六、过渡金属氮化物和硼化物的第一性原理研究金属间化合物相稳定性、层错能及力学性质的第一性原理研究金属间化合物是一类具有广泛应用价值的材料,其在工业、科技、医疗等领域都发挥着重要作用。
然而,金属间化合物的相稳定性、层错能以及力学性质等关键问题一直是科研人员关注的重点。
近年来,随着计算机技术和第一性原理方法的快速发展,对这些问题的研究取得了重要突破。
本文将围绕这三个方面进行详细阐述。
相稳定性是决定金属间化合物能否在实际应用中稳定存在的重要因素。
在高温、高压等极端环境下,金属间化合物的相稳定性更是受到严峻考验。
第一性原理方法能够从原子尺度上揭示金属间化合物的相稳定性规律。
通过计算不同相之间的能量差,可以判断出在特定条件下哪个相更稳定。
还可以通过计算熵、焓等热力学参数,进一步了解金属间化合物在不同环境下的稳定性。
层错能是决定金属间化合物塑性变形能力的重要参数。
在金属间化合物中,层错能的大小直接影响着材料的加工性能和力学性能。
第一性原理方法可以准确地预测金属间化合物的层错能。
通过对比不同材料层错能的差异,可以为材料的加工和优化提供理论指导。
还可以通过调整材料的成分和结构,实现对层错能的有效调控,进一步提高金属间化合物的力学性能。
力学性质是金属间化合物在实际应用中必须考虑的重要因素。
材料的硬度、弹性模量、抗拉强度等都是评价其力学性能的关键指标。
第一性原理方法可以对金属间化合物的力学性质进行全面评估。
通过计算不同应变下的能量变化,可以了解材料的弹性性能;通过模拟裂纹扩展和断裂过程,可以评估材料的韧性和脆性;通过分析原子间的相互作用力,可以预测材料的硬度。
过渡元素的结构化学i
过渡元素,也称为过渡金属,是元素周期表中的一族元素,它们的原子具有未填满的价电子壳层,这使得它们具有独特的化学和物理性质。
在结构化学方面,过渡元素具有一些引人注目的特性。
首先,过渡元素的原子通常具有多个价电子壳层,这意味着它们可以有多个氧化态。
这使得过渡元素在与其他原子或分子相互作用时具有高度的可变性。
例如,铁元素可以在其原子结构中失去两个、三个或全部五个电子,形成二价、三价或五价铁离子。
其次,过渡元素的原子往往具有较低的配位数,这意味着它们倾向于与其他原子形成紧密的结合。
这种紧密的结合是由于过渡金属的空轨道和配位体的电子之间的强烈相互作用。
例如,铜离子倾向于形成四配位的化合物,如硫酸铜。
此外,过渡元素还经常形成具有复杂结构的化合物,如金属间化合物和金属络合物。
这些化合物通常具有特殊的物理和化学性质,可以应用于许多领域,如催化剂、医药和材料科学。
总的来说,过渡元素的结构化学特性使得它们在化学和物理领域中具有广泛的应用。
通过深入研究过渡元素的结构和性质,我们可以更好地了解这些元素的化学行为,从而为未来的科学研究和技术创新提供基础。