16SrDNA的提取&鉴定
- 格式:doc
- 大小:1.60 MB
- 文档页数:11

一、实验原理随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA杂交、rDNA指纹图、质粒图谱和16S rDNA序列分析等。
细菌中包括有三种核糖体RNA,分别为5S rRNA、16S rRNA、23S rRNA,rRNA基因由保守区和可变区组成。
16S rRNA对应于基因组DNA上的一段基因序列称为16S rDNA。
5S rRNA虽易分析,但核苷酸太少,仅几十bp,没有足够的遗传信息用于分类研究;23S rRNA含有的核苷酸数几乎是16S rRNA的两倍,分子量太大,分析较困难。
而16S rRNA相对分子量在2kb左右,较为适合PCR 扩增,又具有保守性和存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16S rRNA作为序列分析对象对微生物进行测序分析。
16SrRNA的编码基因是16SrDNA,但是要直接将16SrRNA提取出来很困难,因为易被广泛存在的RNase降解,因而利用16S rDNA鉴定细菌,其技术路线如下:PCR实验原理即聚合酶链式反应,是指在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。
是一项DNA体外合成放大技术,能快速特异地在体外扩增任何目的DNA。
二、主要器具及试剂PCR、电泳系统、DNA提取体系、Taq Polymerase、DNA Marker,溶菌酶、dNTP和E.coli JM109感受态细胞、pMD18-T Vector、琼脂糖、SDS裂解缓冲液、50×TAE电泳缓冲液贮存液、1×TE(pH 8.0)三、操作方法1. 细菌基因组总DNA的提取接种纯化的菌株于LB液体培养基中,180 r/min,37 ℃培养过夜,按以下的方法提取细菌基因组总DNA。
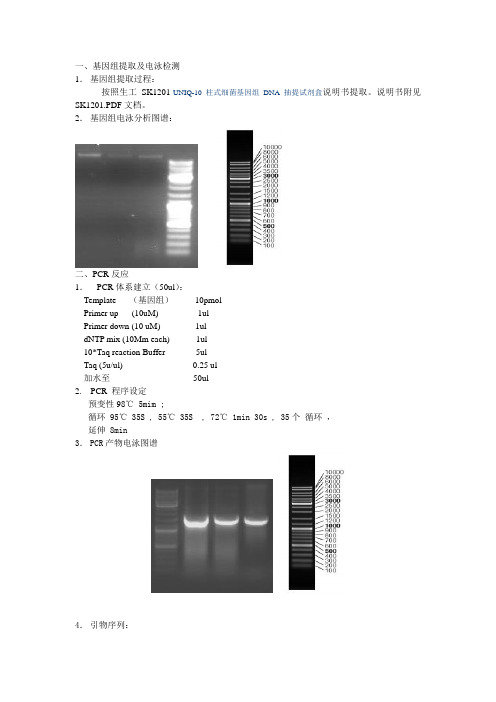
一、基因组提取及电泳检测1.基因组提取过程:按照生工SK1201-UNIQ-10 柱式细菌基因组DNA抽提试剂盒说明书提取。
说明书附见SK1201.PDF文档。
2.基因组电泳分析图谱:二、PCR反应1.PCR体系建立(50ul):Template (基因组)10pmolPrimer up (10uM) 1ulPrimer down (10 uM) 1uldNTP mix (10Mm each) 1ul10*Taq reaction Buffer 5ulTaq (5u/ul) 0.25 ul加水至50ul2.PCR 程序设定预变性98℃ 5mim ;循环 95℃ 35S , 55℃ 35S , 72℃ 1min 30s , 35个循环,延伸 8min3.PCR产物电泳图谱4.引物序列:27f 5’AGAGTTTGA TCCTGGCTCAG 3 ‘ 20bp1492r 5’ GGTTACCTTGTTACGACTT 3’ 19bp三、DNA琼脂糖切胶纯化由PCR产物电泳结果切割所需DNA目的条带,纯化方式见附见说明书SK1131胶回收PDF文档.四、DNA 测序(见测序图谱)五、主要实验仪器PCR反应扩增仪(加拿大BBI公司);3730测序列分析仪( 美国ABI 公司)SW-CJ-1D洁净工作台(江苏苏洁净化设备厂);DK-8D型电热恒温水槽(上海森信实验仪器有限公司);DYY-8型稳压稳流电泳仪(上海琪特分析仪器有限公司);YXJ-2离心机(湘仪离心机仪器有限公司)H6-1微型电泳槽(上海精益有机玻璃制品仪器厂);凝胶成像系统(Gene Genius公司),U-3010紫外-可见分光光度计(Hitachi公司);移液器(范围100-1000μl,20-200μl,0.5-10μl)(加拿大BBI公司);。

16s rdna测序原理16s rDNA测序是一种用于研究微生物群落结构和功能的重要技术,它可以帮助科研人员了解微生物的多样性和相互关系,对环境微生物的研究具有重要意义。
本文将介绍16s rDNA测序的原理及其在微生物学研究中的应用。
16s rDNA是细菌和古细菌的小亚基RNA基因的一部分,它在所有细菌和古细菌中都存在,并且在细菌的进化过程中具有高度的保守性。
因此,通过对16s rDNA序列进行测序和比对,可以帮助科研人员了解不同微生物的分类和演化关系。
在进行16s rDNA测序时,首先需要从样品中提取微生物的DNA,然后通过PCR扩增得到16s rDNA的片段。
接下来,对PCR产物进行纯化和测序准备,最常用的方法是通过测序仪进行Sanger测序。
随着高通量测序技术的发展,现在也可以使用Illumina、454或Ion Torrent等平台进行高通量测序,大大提高了测序的效率和速度。
得到16s rDNA序列后,接下来的工作就是对序列进行比对和分析。
科研人员可以利用公开数据库中的16s rDNA序列作为参考,通过比对来确定待测序列的分类地位和系统发育关系。
此外,还可以利用一些生物信息学工具对序列进行多样性分析、物种丰度分析等,从而了解微生物群落的结构和功能。
在微生物学研究中,16s rDNA测序被广泛应用于环境微生物群落的研究。
通过对土壤、水体、空气等不同环境中微生物的16s rDNA进行测序和分析,可以揭示微生物的多样性、分布规律以及其对环境的影响。
此外,16s rDNA测序还可以用于研究人体内的微生物群落,例如肠道菌群的研究,有助于了解微生物与宿主健康的关系。
总之,16s rDNA测序是一种重要的技术手段,它为科研人员提供了解微生物多样性、分类和系统发育关系的重要途径,对微生物学、生态学和生物医学研究具有重要意义。
随着测序技术的不断发展和完善,相信16s rDNA测序在微生物学研究中的应用将会更加广泛和深入。

一、 实验原理 随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA 杂交、rDNA 指纹图、质粒图谱和16SrDNA 序列分析等。
细菌中包括有三种核糖体RNA ,分别为5SrRNA 、16SrRNA 、23SrRNA ,rRNA 基因由保守区和可变区组成。
16SrRNA 对应于基因组DNA 上的一段基因序列称为16SrDNA 。
5SrRNA 虽易分析,但核苷酸太少,仅几十bp ,没有足够的遗传信息用于分类研究;23SrRNA 含有的核苷酸数几乎是16SrRNA 的两倍,分子量太大,分析较困难。
而16SrRNA 相对分子量在2kb 左右,较为适合PCR 扩增,又具有保守性和存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16SrRNA 作为序列分析对象对微生物进行测序分析。
16SrRNA 的编码基因是16SrDNA ,但是要直接将16SrRNA 提取出来很困难,因为易被广泛存在的RNase 降解,因而利用16SrDNA 鉴定细菌,其技术路线如下:DNA 为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA 互补的子链DNA 的过程。
是一项DNA 体外合成放大技术,能快速特异地在体外扩增任何目的DNA 。
二、主要器具及试剂PCR、电泳系统、DNA提取体系、TaqPolymerase、DNAMarker,溶菌酶、dNTP和感受态细胞、pMD18-TVector、琼脂糖、SDS裂解缓冲液、50×TAE电泳缓冲液贮存液、1×TE()三、操作方法1.细菌基因组总DNA的提取接种纯化的菌株于LB液体培养基中,180r/min,37 ℃培养过夜,按以下的方法提取细菌基因组总DNA。
(1)菌体收集:取新鲜的菌液于EP管中,12000r/min离心30s,弃净上清,收集菌体。

一、实验材料菌种:某浸矿混合菌。
设备:eppendorf管、移液枪、台式高速离心机、电泳仪、水浴锅、PCR仪、无菌操作台、牙签、枪头、酒精灯。
试剂:细菌基因组DNA提取试剂盒、LB液体培养基、LB固体平板培养基、TE 缓冲液(pH 8.0)、切胶回收试剂盒、Taq DNA polymerase,10 mM dNTPmix,引物,双蒸水,琼脂糖、溴乙锭EB、电泳缓冲液(50×TAE电泳缓冲液:取Tris 24.2g,冰醋酸5.7ml,0.25mol/L EDTA(pH8.0)20ml,加蒸馏水至100ml)、pGM-T 克隆试剂盒、Hind III、石蜡。
二、实验步骤1 细菌基因组DNA提取1.1 样品收集菌种培养至OD600为1-1.5,此时1 mL 菌液中约含1.0×109细胞,用灭过菌的1.5mL离心管去菌液1 ml,室温10,000 rpm离心1 min,收集菌体。
1.2 基因组DNA的提取(使用前先在缓冲液GD和漂洗液PW中加入无水乙醇,加入体积参照瓶上的使用说明。
)1.向菌体沉淀中加入200 μl 缓冲液GA,振荡至菌体彻底悬浮;2.向管中加入20 μl 蛋白酶K溶液,混匀;3.加入220 μl 缓冲液GB,振荡15秒,70℃放置10 min,溶液应变清亮,简短离心以去除管盖内壁的水珠;若溶液未变清亮,说明细胞裂解不彻底,可能导致提取DNA量少和提取出的DNA不纯;4.加入无水乙醇,充分振荡混匀15秒,此时可能会出现絮状沉淀,简短离心以去除管盖内壁的水珠;5.将上一步所得溶液和絮状沉淀都加入一个吸附柱CB3中(吸附柱放入收集管中),12,000 rpm离心30秒,倒掉废液,将吸附柱CB3放入收集管中;6.向吸附柱CB3中加入500 μl 缓冲液GD,12,000 rpm离心30秒,倒掉废液,将吸附柱CB3 放入收集管中;7.向吸附柱CB3中加入700 μl 漂洗液PW,12,000 rpm 离心30秒,倒掉废液,吸附柱CB3放入收集管中;8.向吸附柱CB3中加入500 μl 漂洗液PW,12,000 rpm离心30秒,倒掉废液,将吸附柱CB3放入收集管中;9.将吸附柱CB3放回收集管中,12,000 rpm离心2分钟,倒掉废液,将吸附柱CB3置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液;10.将吸附柱CB3转入一个干净的离心管中,向吸附膜的中间部位悬空滴加50-200 μl 洗脱缓冲液TE,室温放置2-5分钟,12,000 rpm 离心2分钟,将溶液收集到离心管中。

一、实验原理随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA杂交、rDNA指纹图、质粒图谱和16S rDNA序列分析等。
细菌中包括有三种核糖体RNA,分别为5S rRNA、16S rRNA、23S rRNA,rRNA基因由保守区和可变区组成。
16S rRNA对应于基因组DNA上的一段基因序列称为16S rDNA。
5S rRNA虽易分析,但核苷酸太少,仅几十bp,没有足够的遗传信息用于分类研究;23S rRNA含有的核苷酸数几乎是16S rRNA的两倍,分子量太大,分析较困难。
而16S rRNA相对分子量在2kb左右,较为适合PCR 扩增,又具有保守性和存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16S rRNA作为序列分析对象对微生物进行测序分析。
16SrRNA的编码基因是16SrDNA,但是要直接将16SrRNA提取出来很困难,因为易被广泛存在的RNase降解,因而利用16S rDNA鉴定细菌,其技术路线如下:PCR实验原理即聚合酶链式反应,是指在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。
是一项DNA体外合成放大技术,能快速特异地在体外扩增任何目的DNA。
二、主要器具及试剂PCR、电泳系统、DNA提取体系、Taq Polymerase、DNA Marker,溶菌酶、dNTP和E.coli JM109感受态细胞、pMD18-T Vector、琼脂糖、SDS裂解缓冲液、50×TAE电泳缓冲液贮存液、1×TE(pH 8.0)三、操作方法1. 细菌基因组总DNA的提取接种纯化的菌株于LB液体培养基中,180 r/min,37 ℃培养过夜,按以下的方法提取细菌基因组总DNA。
16s rdna测序原理16S rDNA测序是一种常用的微生物多样性分析方法,通过对16S rDNA基因的测序和分析,可以揭示微生物群落的组成和结构,对环境微生物的研究具有重要意义。
本文将介绍16S rDNA测序的原理及相关内容。
1. 16S rDNA基因简介。
16S rDNA是细菌和古细菌的小亚基核糖体RNA基因,其序列在细菌中高度保守,但又存在一定的变异性,这使得16S rDNA成为研究微生物系统发育和分类的理想分子标记。
在细菌和古细菌中,16S rDNA一般由9个高度保守的区域(称为conserved region)和10个变异区域(称为variable region)组成,其中变异区域的序列差异较大,可用于微生物的分类和鉴定。
2. 16S rDNA测序原理。
16S rDNA测序的原理是通过PCR扩增获得16S rDNA基因片段,然后对扩增产物进行测序,最后对测序结果进行分析和解读。
首先,从样品中提取微生物DNA,然后利用通用或特异引物对16S rDNA基因进行PCR扩增,得到所需的16S rDNA片段。
接下来,对PCR产物进行纯化和测序,通常采用Sanger测序法或高通量测序技术(如Illumina、454、Ion Torrent等)。
最后,利用生物信息学方法对测序结果进行分析,包括序列比对、物种注释、多样性分析等。
3. 16S rDNA测序分析。
在16S rDNA测序分析中,首先需要对测序结果进行质控和过滤,去除低质量序列和引物污染,然后进行序列比对和物种注释。
序列比对是将测序结果与16S rDNA数据库进行比对,找到最佳匹配的参考序列,从而确定微生物的分类和系统发育关系。
物种注释是根据比对结果,将未知序列注释为已知的微生物分类单元,如属、种等。
此外,还可以进行多样性分析,如Alpha多样性指数(反映微生物群落的丰富度和多样性)、Beta多样性分析(比较不同样品间的微生物群落差异)等。
细菌16SrDNA测序鉴定一、细菌总DNA提取二、16SrDNA的扩增1、以单菌落或菌悬液为模板的16SrDNA扩增体系25ul体系Buffer(含有Mg2+):2.5 uldNTP:2.5 ul:3,-primer:0.5ul5,-primer:0.5ul模板:1个单菌落(或1 ul在LB培养液中37℃,12h的菌悬液)吐温20:2ulTaq酶:0.3水:16.7(或15.7ul)细菌16S rDNA的通用引物为Pf-AGAGTTTGATCCTGGCTCAG和Rf-TACCTTGTTACCACTT许敬亮博士论文66页:扩增采用通用的引物,其正向序列为:5'-AGAGTTTGATCCTGGCTCAG-3',反向互补序列为: 5'-TTCAGCATTGTTCCAT-3'。
黄婷婷博士论文70页:5’端:5'-AGAGTTTGATCCTGGCTCAG-3'(Escherichia colibases 8 to 27),3’端:5'-TACCTTGTTACGACTT-3'(Escherichia coli bases 1507 to 1492)。
2、以总DNA为模板的16SrDNA扩增体系25ul体系Buffer(含有Mg2+):2.5 ul (购买)dNTP:2.5 ul:3,-primer:0.5ul5,-primer:0.5ul模板:1 ulTaq酶:0.3(或0.2ul)双蒸水:17.7(或17.8ul)3、PCR反应条件94℃,5min;95℃,变性30s;50℃~52℃(可根据扩增情况进行适当调节),退火30s;72℃,延伸1min;30个循环;72℃,延伸15min;10℃(or4℃),保温10min(or nh)。
4、检测扩增效果取1~3 ul扩增产物,适量load buffer,以DL2000为Marker,上0.75%琼脂糖凝胶跑电泳(电压挑低些80~100,胶长些,利于各扩增产物分离及回收切胶)注:扩增条件摸索时每个模板只扩1管即可三、16SrDNA的回收扩增出的每一管16SrDNA经0.75%琼脂糖凝胶电泳检测后,将效果好的各管16SrDNA合并使达到60~100ul样量,100ul16SrDNA+10ul溴酚蓝跑电泳回收胶(电泳液需换成新鲜的,DL2000为Marker),EB染色后在紫外灯下切下16SrDNA条带放入1.5ml离心管中(离心管事先称重),按DNA凝胶回收试剂盒操作说明回收16SrDNA,取回收16SrDNA 2ul(Dl2000为Marker)跑电泳观察浓度(亮度)与纯度(条带数与位置)。
海润德饲用微生物菌种的16SrDNA分子鉴定技术路线一、海润德饲用微生物菌种的16SrDNA分子鉴定技术路线↓提取分离菌株基因组DNA↓以细菌基因组DNA为模板,P1、P2为引物,PCR扩增其16SrRNA↓PCR产物切胶回收↓连接PMD18T-vector↓转化Ecoli DH5а;蓝白斑初筛、PCR、酶切鉴定重组质粒↓将鉴定正确的重组质粒,送交测序↓测序结果在NCBI上BLAST分析二、试验方法1、细菌基因组提取方法方法一:细菌基因组简易提取方法(参考国外文献)1.取1.5ml细菌过夜培养物,12000rpm,离心0.5min,弃上清。
2.加入200ul lysis buffer( 40 mM Tris-Cl; 20Mm NaAC; 1Mm EDTA; 1%SDS.),剧烈振荡。
(移液枪反复吹打或者涡漩,目的使菌体充分悬浮)。
3.加入66ul 5M NaCl剧烈混合。
4.离心12000rpm,10 min 取上清。
5.加入等量氯仿:异戊醇(24:1),反复颠倒50次混匀.(呈牛奶色)。
6.离心12000 rpm ,10min,取上清。
7.加2倍的无水乙醇,置-20℃冰箱,沉淀DNA。
8.加适量70%乙醇漂洗沉淀9.DNA与超净台,风干。
10 加适量ddH2O溶解DNA。
11.加0.5-1 ul RNAase,37℃作用20 min 。
12.-20℃冰箱保存。
方法二:SDS/CTAB法1.取1.5ml细菌过夜培养物,12000rpm,离心0.5min,弃上清。
2.加500ul TE 缓冲液充分悬浮菌体。
3.加30μl 10%SDS,3μL 蛋白酶K,混匀,37℃,1h.4.加 100μl NaCL(5M) 混匀。
5.加80μl 的CTAB/NaCL,混匀;65℃ 10min 。
6.加入等体积酚/氯仿/异戊醇混合液,混匀。
7.离心12000rpm,4-5min8.重复6、7步骤,抽提2-3次9.12000 rpm ,离心10min,取上清。
16SrDNA-DGGE技术基本路线一样品总DNA的提取1 实验器材:玻璃器材,铝盖,1.5mL离心管,螺口管,锆珠,搅拌器,4℃离心机。
2 实验试剂:PBS(0.05M,pH=7),水饱和酚,TE饱和酚,TN150,TE缓冲液,TAE缓冲液,NaAc (pH=5.2),氯仿/异戊醇(24:1)(V/V),96%冷乙醇,70%冷乙醇。
3 实验操作:3.1 细胞分裂称取0.5g样品入含0.3g锆珠、已灭菌的screw-capped tube 中,加入1mL TN150,和150µL酸性酚,bead-beater匀浆(5000rpm,3min),冰上冷却,加入150µL氯仿/异戊醇,涡旋混匀,10000rpm离心,5min转移上清液至已灭菌的eppendorf管3.2 DNA提取加入150µL氯仿/异戊醇,和150µLTE饱和酚至上述eppendorf管中涡旋混匀1min,10000rpm离心1min,上清移入灭菌的eppendorf管中,重复上述步骤直至水相之间的界面清晰为止,上清移入灭菌的eppendorf管中,加入300µL氯仿/异戊醇,10000rpm离心1min,上清移入灭菌的eppendorf管中,加入96%冷乙醇1mL,和50µL3M NaAc (pH=5.2),-20℃沉淀DNA过夜。
10000rpm离心20min弃上清,加入500µL70%冷乙醇溶解沉淀,10000rpm离心5min弃上清。
风干沉淀加入50µLTE缓冲液溶解DNA-20℃保存提取的DNA4实验注意事项:酚是致癌物质,实验操作应配带手套,且不要将该溶液洒在桌面上或地面上。
实验中使用了挥发性有毒物质,操作应在通风橱中进行。
实验过程中样品应放置冰面上,所有离心操作在4℃下进行。
二DNA的检测—琼脂糖凝胶电泳1试剂和器材:电泳缓冲液(1*TAE),溴乙锭(EB)染色贮存液(1mg/mL),0.25%溴酚兰,琼脂糖,双蒸水。
用16S rDNA 方法鉴定细菌种属一、实验目的1. 掌握16S rDNA 对细菌进行分类的原理及方法;2. 掌握DNA 提取、PCR 原理及方法、DNA 片段回收等实验操作。
二、实验原理随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA 杂交、rDNA 指纹图、质粒图谱和16S rDNA 序列分析等。
细菌中包括有三种核糖体RNA ,分别为5S rRNA 、16S rRNA 、23S rRNA ,rRNA 基因由保守区和可变区组成。
16S rRNA 对应于基因组DNA 上的一段基因序列称为16S rDNA 。
5S rRNA 虽易分析,但核苷酸太少,没有足够的遗传信息用于分类研究;23S rRNA 含有的核苷酸数几乎是16S rRNA 的两倍,分析较困难。
而16S rRNA 相对分子量适中,又具有保守性和存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16S rRNA 作为序列分析对象对微生物进行测序分析。
利用16S rDNA 鉴定细菌的技术路线:PCR 的基本原理:必须已知部分序列设计出引物,10-30bp 。
细菌培养液 基因组DNA DNA 提取 PCR 扩增 16S rDNA 片段片段回收 连接克隆载体 阳性克隆鉴定 测 序 序列比对以此类推,呈几何级增长。
一般进行25-35个扩增循环DNA可扩增106~109倍。
三、实验步骤(一)细菌基因组DNA提取技术路线:具体步骤:1. 挑单菌落接种到10 mL LB培养基中37℃振荡过夜培养。
(前期由老师完成)2. 取2 mL培养液到2 mL Eppendorf管中,8000 rpm离心2分钟后倒掉上清液。
3. 再往同一离心管中2 mL培养液到2 mL Eppendorf管中,8000 rpm离心2分钟后倒掉上清液。
4. 加入200 μL 缓冲液GA,充分悬浮、混匀,将菌体彻底悬浮。
5. 再加入20 μL Protein K,混匀,55℃温育10分钟。
(55℃为酶最适温度)6. 加入220 μL缓冲液GB,混匀后,55℃温育10分钟。
(实为50-60℃之间)7. 加入220 μL 无水乙醇(降低体系粘稠度),充分混匀。
将上清(挑去其中絮状杂质,少部分,会产生气泡,除不去))小心转入UNIQ-10柱(羟基纤维素膜,特异吸附核酸,因为吸附有限,所以损失核酸)中。
13000 rpm(离心机最高设定实为10000rpm)离心5分钟,倒弃收集管内的液体。
6. 加入500 μL GD Solution,10000 rpm离心1分钟。
8. 加入500 μL 70%乙醇(Wash Solution),10000 rpm离心0.5分钟。
(离心机最小设定实为1min,仪表盘上显示时间的梯形最小面积为1min,所以没法在到梯形面积中间时按暂停)9. 再10000 rpm离心2分钟彻底甩干乙醇。
吸附柱转移到一个新的1.5 mL的离心管。
10. 加入50 μL预热(60℃)的洗脱缓冲液(TE,含RNase),加到柱子中间的膜上。
60℃放置5分钟。
12000 rpm离心2分钟,流下的液体即为基因组DNA。
11. 电泳。
取5 μL提取DNA溶液+2μL Loading Buffer(不用吸放混匀,以免机械损伤DNA)进行电泳检测质量。
(吖啶橙做染色液,橙红色)(二)PCR扩增1. 根据已发表的16S rDNA序列设计保守的扩增引物。
16S (F) 5'-AGAGTTTGATCCTGGCTCAG-3'16S (R) 5'-GGTTACCTTGTTACGACTT-3'2. PCR扩增体系:在0.2 mL Eppendorf管中加入1μL DNA,再加入以下反应混合液:16S (F) 1μL (10μM)16S (R) 1μL (10μM)2×PCR Mix 25 μL(含10×PCR Buffer,dNTP,Taq酶)ddH2O22 μL(最先加)反应体系50 μL,简单离心混匀。
3. PCR反应将Eppendorf管放入PCR仪,盖好盖子,调好扩增条件。
扩增条件为:94℃ 3 min94℃30 sec50℃(实为52℃)45 sec 35 cycles72℃100 sec(因为Taq酶1Kb/min,时间为经验所得)72℃7 min(充分延伸)4. PCR产物的电泳检测拿出Eppendorf管,取出全部溶液点入预先制备好的1%的琼脂糖凝胶大孔中。
电泳1 hr。
在紫外灯下检测扩增结果(下午1:50)。
(三)扩增片段的回收根据上步实验结果,如果扩增产物为唯一条带,可直接回收产物。
否则从琼脂糖凝胶中切割核酸条带,并回收目的片段。
1)称量一2 mL.的Eppendorf 管质量,记录0.99g 。
2)在紫外灯下切割含目的条带的凝胶,放入2 mL 的Eppendorf 管内,称量1,06g 。
计算凝胶质量0.07g 。
3)每100 mg 凝胶加入300 μL Buffer DE-A (促融),混匀。
70℃温育至凝胶融化。
4)加入0.5倍体积的Buffer DE-B (帮助连接),混匀。
全部转入UNIQ-10柱,10000 rpm 离心1分钟,倒去收集管(与之前的试剂盒中的收集管规格不同)内的液体。
5)加入500 μL Buffer W1(Wash Solution ),10000 rpm 离心1分钟,倒去收集管内的液体。
6)加入500 μL Buffer W2(去盐液,70%乙醇),10000 rpm 离心0.5分钟。
(离心机最小设定实为1min )7)再10000 rpm 离心2分钟彻底甩干乙醇。
吸附柱转移到一个新的1.5ml 的离心管。
8)加入30 μL 预热的洗脱缓冲液(Eluent ),60℃放置3分钟。
12000 rpm 离心2分钟,流下的液体即为回收的DNA 片段。
(四)DNA 片段测序将回收的片段(足量)送至生物公司测序,测序引物为16S PCR 引物。
(Sanger 双脱氧法,一边PCR ,一边测序)四、实验结果及分析1.细菌基因组提取DNA 电泳结果:第十一泳道为我组结果,条带清晰,说明提取的DNA 含量较高。
正极 负极 点样孔(小孔,4ul )DNA28sRNA18sRNA5.8sRNA2. PCR产物的电泳检测:负极正极点样孔为大孔,96μL。
Marker 6条带,清晰明亮。
第五泳道为我组结果,可见条带清晰,说明提取的DNA含量较高。
3. 根据测序结果,到NCBI Blast项目上进行比对,确定该未知菌的种属。
分析讨论实验的过程及结果。
选择序列三:1)打开网站,选取BLAST。
2)选择核酸比对3)将序列粘贴到框中,下面的方框选Reference genomic sequence,按BLAST,得到结果。
序列三与葡萄球菌属细菌的16S rDNA基因相同性最高,与Staphylococcus saprophyticus subspATCC.的16S rDNA基因的相同性为99%。
可初步判定序列三所属菌株可能是Staphylococcus属的成员。
如果要明确的得到其种属,还要进行相关的生理生化分析。
(四)结果处理:生成文件,制作进化树。
1)点击Rrna-16S ribosomal RNA进去后,按图示点击,Create Files,保存在文件夹中,供生成进化树时使用。
每一种保存一次,我们共对20组生成了文件,保存在一个文档中。
2)运行MEGA 5.05软件,按如下操作,打开下载下来的Sequence文件。
4)点击Alignment,Align by ClustalW,然后按OK键,等待。
5)得到以下画面,删除序列两端不能对齐的碱基(不同源)。
6)按Data,Phylogenetic Analysis,关闭窗口,将文件保存。
7)选择一个方法,构建系统发育树。
8)Bootstrap 选择1000,点击compute开始计算。
9)得到最终的进化树。
(我们用的是样品3,命名为Alice)五、分析讨论(一)本次试验主要是通过16S rDNA的方法来鉴定细菌种属,通过提取基因组DNA、PCR扩增、电泳检测,最后将得到的序列在NCBI上比对,找出与之同源性最大的菌种,最后用MEGA4软件制作出进化树。
在做这个时要注意的地方:①细菌基因组DNA的提取中,加入缓冲液GA后要彻底悬浮菌体,来回吸打几次,使菌体彻底悬浮,切记不要产生气泡。
②加完GB缓冲液后,手拿Eppendorf管轻轻地上下颠倒混匀,令其充分反应,破坏细胞膜。
加完无水乙醇后会看到澄清透亮的液体中有一团白色的残渣漂浮着,用枪头小心地把它挑出,避免抽入不溶性的絮状沉淀,否则后面会出现较多的粘稠状物质堵住膜孔③离心前要精确平衡,避免对离心机造成机械损害④预热的洗脱缓冲液要竖直打到硝酸纤维素膜上,该膜能特异性的吸附核酸,若打到壁上会造成损失⑤提取好的基因组DNA要在4℃条件下保存⑥在制作PCR扩增产物时,先加水,打DNA液时将枪头伸入水中,防止有DNA液损失在壁上。
其他的可以几个人一组制成大体系混合液,这样取用更加方便而且准确;⑦取液时注意试剂瓶上的标志,如10×、6×等,用前记得稀释;⑧割胶的时候要带手套,带好紫外线防护罩,防止伤害眼睛。
⑨通过查找资料,我们摸索出了在NCBI上进行序列比对以及构建进化树的方法,过程很艰辛,但结果令人欣慰。
⑩构建进化树时,一定要参考多和同学们讨论,有条件时多多向实验室的研究生学长学姐请教,他们经常会用到BLAST和MEGA软件,知道很多小技巧和注意事项,在做进化树的时候给我很大的帮助。
⑪(二)1.由于核糖体进化保守,所以可以通过测定核糖体16S rDNA(序列约1.5KB)来鉴定细菌种属。
若要鉴定真核生物,则测定核糖体18S rDNA。
2.氯仿抽提可以去除蛋白(中间层)和有机物,G+菌用溶菌酶,G-菌用细胞膜裂解液。
GA为Tris-Hcl,GB为裂解缓冲液SDS。
3.好引物的特点:扩增产物多,杂带少。
4.由于分子生物学实验的特点是实验时间长,中间等待时间长,涉及的高值仪器多,接触有害试剂多,实验原理较复杂,所以我们要严格遵守实验室纪律,认真做好实验,认真做好值日,从有限的学时中真正地学到东西。