TCGA癌症数据库介绍专题
- 格式:pptx
- 大小:1.97 MB
- 文档页数:29

tcga 基因水平拷贝数-回复题目:TCGA基因水平拷贝数分析:基于大规模癌症基因组数据的解读引言:近年来,基因组学研究在癌症领域取得了巨大的突破,其中TCGA(The Cancer Genome Atlas)项目收集了大规模的癌症基因组数据,为我们深入理解癌症发生机制提供了重要的资源。
在TCGA数据库中,基因水平拷贝数作为一种常见形式的基因组变异,具有关键的生物学意义。
本文将一步一步回答TCGA基因水平拷贝数相关问题,分析其特征、检测方法以及与癌症发生关系的研究进展。
一、基因水平拷贝数:概述与特征1.1 基因水平拷贝数的定义基因水平拷贝数(gene-level copy number)指的是基因组中各个基因的拷贝数变异情况。
拷贝数变异是一个细胞的基因组拷贝数与正常人群的拷贝数之间的差异。
正常情况下,每个基因通常有两个拷贝数(一个来自父本,一个来自母本),然而在某些情况下,基因的拷贝数可能会增加或减少,从而导致拷贝数变异现象。
1.2 基因水平拷贝数的特征基因水平拷贝数的特征可以通过TCGA数据库的大规模基因组数据进行分析得到。
常见的基因拷贝数变异现象包括基因扩增(基因拷贝数增加)、基因缺失(基因拷贝数减少)以及染色体局部的拷贝数增加或减少。
这些拷贝数变异通常与癌症的发生和发展密切相关。
二、TCGA基因水平拷贝数数据分析方法2.1 TCGA数据库介绍TCGA项目收集了多种肿瘤类型的癌症患者样本,包括肿瘤组织和正常对照组织。
通过测序技术和芯片技术,TCGA数据库提供了大量的基因组数据,包括基因水平拷贝数数据。
2.2 TCGA基因水平拷贝数数据获取TCGA数据库提供了公开获取基因水平拷贝数数据的功能,用户可以通过访问TCGA官方网站或者特定的数据库平台(如UCSC Xena)来下载感兴趣的数据。
2.3 TCGA基因水平拷贝数数据预处理为了获得可信的结果,TCGA基因水平拷贝数数据需要经过预处理步骤,如数据质量控制、均值中心化、标准化等。

Comprehensive Characterization of Cancer Driver Genesand MutationsCell, April 2018背景●在精准肿瘤学中,识别癌症驱动分子至关重要。
●虽然目前存在一些算法来识别驱动事件,但缺乏将这些算法整合、优化、并应用于大数据中的研究。
●本研究对33种疾病类型、9000多个患者的致癌驱动基因和突变进行最大规模的系统性研究,并强调了在肿瘤患者中普遍存在一些临床可诉性驱动事件。
摘要●对TCGA数据库中33个癌种、9423例患者的外显子组采用PanSoftware 策略分析(共涉及到26个计算工具),来识别驱动基因和驱动突变事件。
●共识别了299个驱动基因、这些基因与它们的解剖学位点及癌症/细胞类型相关。
●基于序列和结构的分析,识别了>3,400个错义驱动突变位点,这些位点被多线证据支持。
●预测到的驱动突变中,有60%~85%的位点通过了实验验证。
●300多个MSI的肿瘤样本与PD-1/PD-L1的高表达相关,其中有57%的肿瘤样本存在临床可诉性事件。
意义该研究是迄今为止,对癌症基因和突变进行的最大规模的系统性研究,可以为将来的生物学和临床研究做出指导。
1. 癌症驱动基因的识别策略与效能●Figure 1A. 发现癌症驱动基因的策略:数据收集、工具开发、异常值校正、人工搜索、下游分析以及功能验证。
●Figure 1B. 在每个癌症类型中,体细胞突变数目均成广泛分布。
●Figure 1C. 每个癌种中,6种碱基的类型改变分布(转换和颠换)。
●Figure 1D. 对于单个癌症,有效突变的背景突变率中值为6.1%,且样本量越大,统计效能越好。
2. 癌症驱动基因的识别1.共识别了299个基因,其中利用系统方法识别到了258个基因,41个基因是利用人工搜索及额外的组学工具发现的。
2.Figure 2A. 外层每个扇形代表一个癌种,以及该癌种特异突变的驱动基因。

癌症和肿瘤基因图谱(TCGA)计划简介据统计,全球每年新增癌症患者达700万人,死于癌症的病人达500万人,60%的患者确诊后只能存活5年。
目前已知的癌症有200多种,但是,无论什么癌症,在肿瘤的特殊类别(分型)或发展的不同分期方面都发现有基因组的特异变化,而正是基因组的改变(突变)导致了细胞分化、发育和生长通路的不正常,从而引发细胞不正常地失控增殖、生长。
美国政府发起的癌症和肿瘤基因图谱(Cancer Genome Atlas,TCGA)计划,试图通过应用基因组分析技术,特别是采用大规模的基因组测序,将人类全部癌症(近期目标为50种包括亚型在内的肿瘤)的基因组变异图谱绘制出来,并进行系统分析,旨在找到所有致癌和抑癌基因的微小变异,了解癌细胞发生、发展的机制,在此基础上取得新的诊断和治疗方法,最后可以勾画出整个新型“预防癌症的策略”。
2005年12月13日,这一项目由美国国家癌症和肿瘤研究所(NCI)和国家人类基因组研究所(NHGRI)联合进行,预计耗资1亿美元。
和人类基因组计划(HGP)相似,TCGA是另一项以基因组为基础的大科学研究计划,它以人类基因组计划的成果为基础,研究癌症中基因组的变化。
与HGP专注于疾病的遗传因素(与生俱来)不同,TCGA更关心人类出生后细胞中的基因变化(后天变异)。
大部分癌症在威胁到健康之前都会产生几种体细胞突变(somatic mutations),而这些所谓的体细胞或获得性突变是不可遗传的。
TCGA 是迄今为止世界上所进行的最大一项基因工程,差不多能抵上100多个HGP,在3年探索初期就要绘制出比HGP更多的基因图谱。
绘制癌症基因图谱有助于把研究人员从目前逐个追踪基因的大量劳动中解放出来,便于迅速设计和找到针对性抗癌药物。
美国国家癌症研究所副所长安娜•巴克认为,这项计划“是生物医学研究中的一大转折点,也是药物治疗的一大转折点”。
国立卫生院主管John E. Niederhube医学博士说道“今天我们得到一种新的观点去审视遗传改变在一生当中的蓄积与恶性肿瘤的联系。
1.从TCGA下载相应的癌症数据,包括正常样品和癌症样品。
2.差异的lncRNA和microRNA分析。
分别对mRNA和miRNA做差异表达分析,得到差异的miRNA和基因,并绘制热图和火山图。
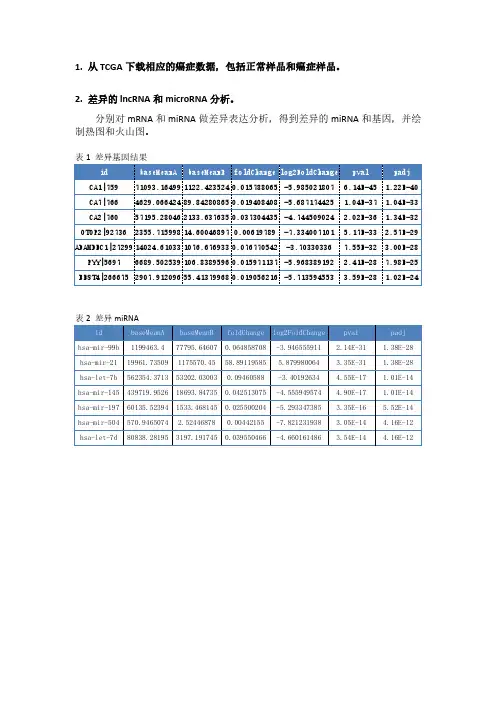
表差异
图1差异基因火山图
图2热图
3.共表达网络
基因和miRNA的共表达网络。
图3miRNA和mRNA共表达网络4.蛋白互作网络
对差异基因进行蛋白互作网络分析。
图4差异基因PPI网络
5.生存分析
分析基因高低表达与生存时间之间是否具有显著相关性,并且绘制生存曲线。
当然,也可以分析临床信息与生存的关系,比如临床分期与生存时间的关系,癌症大小与生存时间的关系,用药与生存时间的关系,等等。
图5目标基因生存分析
6.基因表达与临床的关系
分析基因与临床数据的关系,如基因的表达和癌症转移的关系,基因表达和临床分期的关系,基因表达和其它临床信息的关系。
图6MARCH1表达与肺转移的关系
7.其它个性化分析
根据客户提供分析案例或者文献,做相应的生物信息分析。
有疑问请联系作者邮箱:602316645@。

基于TCGA的肺腺癌组织中CDT1表达及相关信号通路分析1,1,许培培21武汉大学中南医院,武汉430071;2郑州大学第三附属医院摘要:目的基于症基因组图谱(TCGA)数据库,观察染色质许可和DNA复制因子1(CDT1)在肺腺癌患者癌组织中的表达变化,分析癌组织中CDT1与患者临床病理特征、预后的相关性,并预测CDT1在肺腺癌中参与调节的信号通路。
方法从TCGA数据库中下载肺腺癌组织中CDT1基因表达谱及肺腺癌患者的临床病理信息,使用R3.6.1软件提取肺腺癌组织(肺腺癌组)及正常肺组织(正常对照组)CDT1的表达量数据,利用Mann-Whitney U 检验比较两组间的表达差异。
以CDT1表达水平的中位值(4.409)为界限将肺腺癌患者分为CDT1高表达组和CDT1低表达组,利用单因素及多因素COX回归分析癌组织中CDT1表达与患者临床病理特征的关系。
利用R3.6.1软件"survival"包分析CDT1高、低表达组总体生存率(OS)的差异,并通过GEPIA、Kaplan Meier-plotter、UAL-CAN等在具对结果进行验证。
利用基因分析(GSEA)预测CDT1在肺腺癌中参与的分子通路。
结果肺腺癌组CDT1的表达水平高于正常对照组(P<0.001)。
肺腺癌组织中CDT1的表达水平与患者年龄(P=0.019)-.Stage分期(P=0.007)及远处转移(P=0.042)等相关。
Stage分期(HR=1.97,95%CI: 1.22~3.17,P=0.005)和CDT1表达(HR=1.43,95%CI: 1.14~1.79,P=0.002)可以作为肺腺癌的独立预后因素。
CDT1高表达组生存率低于CDT1低表达组(P=0.029)o CDT1主要参与细胞周期、瞟吟与疇睫代谢、核Z酸切除修复以及p53信号通路等。
结论CDT1在肺腺癌患者中高表达,与肺腺癌患者的Stage分期及远处转移等相关,可作为导癌不良预后的独立危险因子,并通过参与多种信号通路促进肺腺癌的发生发展。

基于整合的TCGA数据库探索基因组学与临床数据关系一、本文概述随着生物信息学和临床研究的不断深入,基因组学与临床数据之间的关联日益成为生物医学领域的研究热点。
本文旨在通过整合和分析公开的The Cancer Genome Atlas(TCGA)数据库,探索基因组学与临床数据之间的关系。
我们将系统介绍如何利用TCGA数据库的资源,运用生物信息学方法,挖掘基因组学数据中的潜在信息,并与临床数据进行整合分析,以期揭示癌症发生、发展过程中的关键基因和分子机制,为癌症的诊断、治疗和预后评估提供新的思路和方法。
本文将首先介绍TCGA数据库的概况和数据特点,阐述选择TCGA 数据库作为研究基础的原因。
随后,我们将详细介绍基因组学数据的处理方法,包括数据清洗、基因表达分析、基因变异检测等,并阐述如何将这些方法与临床数据进行有效整合。
在结果展示部分,我们将通过图表和统计分析,展示基因组学与临床数据之间的关联,并解释这些关联在癌症研究中的意义。
我们将讨论本文的局限性,并对未来的研究方向进行展望。
通过本文的研究,我们期望能够为深入理解癌症的基因组学特征和临床表型提供新的视角和工具,为癌症的精准医疗提供科学支持。
我们也希望本文的研究方法和结果能够为其他领域的生物医学研究提供借鉴和参考。
二、TCGA数据库概述The Cancer Genome Atlas (TCGA) 是一个由美国国家癌症研究所(NCI)和国家人类基因组研究所(NHGRI)共同发起的项目,旨在通过应用高通量的基因组测序技术,对多种类型的人类癌症进行深入的基因组学研究。
自2006年启动以来,TCGA已经产生了海量的多维度数据,包括基因组、转录组、表观组、蛋白质组以及临床数据等,涵盖了超过33种不同类型的癌症,总计数千个患者的样本。
TCGA数据库不仅提供了丰富的原始测序数据,还通过严格的数据处理和分析流程,生成了大量的二级和三级数据,如基因变异注释、基因表达量统计、生存分析等。

TCGA数据库的利用(三)—做差异分析的三种方法做差异分析是TCGA数据库中常见的一项分析任务,可以用来对比两个或多个样本、组织或条件之间的差异,帮助研究人员发现与特定疾病相关的基因或基因组变化。
在TCGA数据库中,常用的差异分析方法包括聚类分析、差异表达基因分析和通路分析。
聚类分析是一种将样本根据基因或基因组数据的相似性进行分组的方法。
这种方法可以帮助研究人员发现基于基因表达的分子亚型和样本亚群。
在TCGA数据库中,研究人员可以选择感兴趣的基因或基因组范围,并将样本进行无监督聚类分析。
通过对聚类结果进行可视化和分析,研究人员可以发现基因或基因组的差异表达模式,并研究其与疾病相关性。
差异表达基因分析是一种比较两个或多个组之间基因表达差异的方法。
在TCGA数据库中,研究人员可以选择感兴趣的组别,比如癌症样本和正常样本,然后使用差异表达基因分析来鉴定不同组别之间的基因表达差异。
差异表达基因分析可以通过一系列统计方法和假设检验来确定哪些基因在不同组别之间的表达存在显著差异。
研究人员可以利用这些差异表达基因,进一步研究其在特定疾病中的生物学功能和作用机制。
通路分析是一种基于差异表达基因或差异基因组的生物学通路富集分析方法。
通过将差异表达基因或差异基因组映射到已知的生物学通路数据库,可以发现在特定疾病中受影响的通路集合。
通路分析可以帮助研究人员理解基因或基因组变化对疾病发生和发展的影响,以及潜在的治疗靶点和生物标记物。
综上所述,利用TCGA数据库进行差异分析可以帮助研究人员发现与特定疾病相关的基因或基因组变化。
聚类分析可以帮助发现基因或基因组的差异表达模式和样本亚群;差异表达基因分析可以确定不同组别之间的基因表达差异;通路分析可以发现受影响的生物学通路。
这些方法可以在研究人员深入探索特定疾病的发病机制和寻找潜在治疗靶点方面起到重要的作用。

手把手教学:轻松玩转TCGA何为TCGA?人类基因组计划(HGP)完成后,癌症研究也早已步入基因组学时代,多维、海量数据产生的速度远远超过理解、分析、处理数据的速度。
在这样的大背景下,美国国家癌症研究院(NCI)和美国国家人类基因组研究院(NHGRI)于2005年发起TCGA (TheCancerGenomeAtlas)项目。
和人类基因组计划(HGP)相似,TCGA是另一项以基因组为基础的大科学研究计划,它以人类基因组计划的成果为基础,研究癌症中基因组的变化。
与HGP专注于疾病的遗传因素(与生俱来)不同,TCGA更关心人类出生后细胞中的基因变化(后天变异)。
图为NBCI历年“TCGA”相关文章数量TCGA数据库包含11,000个病人的33种肿瘤的7个不同层面的基因数据(包括基因表达、CNV,SNP,DNA甲基化,miRNA,外显子组等)和临床数据,意在解析癌症发生的分子机制、肿瘤的亚型和治疗靶点等。
TCGA中的数据可谓包罗万象,常见的有转录组(RNASeq或表达谱芯片)、基因组(外显子或全基因组测序)、表观遗传(甲基化芯片)、蛋白组等多组学数据,最重要的,TCGA中的每个样本都有丰富、准确的临床数据,包括生存时间、肿瘤分期、病理类型等重要临床信息显著优于其他肿瘤数据库。
我们知道,肿瘤的发生与基因突变有很大关系,相关基因的点突变、小片段缺失和插入,引起了密码子的同义、错义、终止和移码的突变现象,导致基因表达的蛋白质由于序列的改变使其相关功能丧失,最终引发细胞的恶变与增殖,产生肿瘤。
太多太多的文章都在研究和肿瘤相关的驱动基因(drivergene)或者体细胞突变(somaticmutation),试图分析基因突变与肿瘤发生发展之间的相关性。
以往的研究中,我们可能要沿着“收集样本-DNA抽提-建库测序-数据分析”这一流程从头到尾走一遍,找几个人合作,再花个几年时间摸索,等到花都谢了,才能得到最终的结果。


基于TCGA数据库胃癌LincRNA生物标志物筛选及生物学功能分析摘要:目的:预测与胃癌相关的LincRNA,及与其相关的信号通路,并筛选出特定的LincRNA作为胃癌预后潜在的生物标志物。
方法:第一步,从TCGA(The Cancer Genome Atlas,TCGA)数据库中下载375例胃腺癌组织和32例癌旁组织的RNA-seq数据(HTseq-RNA Counts)及胃癌患者相关临床信息,并分别提取相关的LincRNA和mRNA。
第二步,使用R语言/Bioconductor的edgeR包分别筛选出差异表达的LincRNA、mRNA。
第三步,使用加权基因共表达网络分析(WGCNA)方法对数据进行加权,得到与胃癌临床特征相关性最大的模块及相对应的临床特征。
第四步,通过CytoHubba 软件寻找关键基因,得到与胃癌发生相关性最大的关键LincRNA。
第五步,对所得到的关键基因LincRNA进行靶基因预测及生物学功能分析,研究与胃癌发生、转移有关的LincRNA与mRNA的相关性,并使用Cytoscape构建LincRNA与mRNA的共表达网络。
第六步,将得到的胃癌关键LincRNA使用Kaplan-Meier软件进行生存分析,以研究LincRNA与胃癌患者的总体存活之间的关联。
结果:1.此研究筛选出2200个差异表达的LincRNA以及4623个差异表达的mRNA;2.得到与肿瘤分级相关性最大的MEbrown模块、MEturquoise模块(模块中为基因群);3.筛选出LincRNA相关性最大模块中的5个关键基因(Hub基因),分别是BARX1-AS1、CARMN、FENDRR、GAS1RR、LINC01354;4.得到胃癌发病可能相关的MAPK Signaling Pathway、CELL CYCLE Signaling Pathway、P53 Signaling Pathway、P13K-AKT Signaling Pathway、WNT Signaling Pathway、TGF-BETA Signaling Pathway、ADHERENS JUNCTION Signaling Pathway七条主要的KEGG生物信号通路;5.构建LincRNA 与mRNA共表达网络;6.BARX1-AS1、CARMN、FENDRR、GAS1RR、LINC01354高表达组生存率均较低表达组低。

Oncogenic Signaling Pathways in The Cancer Genome AtlasCell, April 2018背景●过去十年,DNA测序使得系统研究肿瘤基因突变成为可能,使得人们对肿瘤的发生过程和相关信号通路有了更深的认识;●肿瘤相关的基因和通路变异数目众多,加深对这些基因和通路变异的认识对开发潜在临床治疗方案十分必要;●前人研究已经发现很多重要的肿瘤相关信号通路,且有TCGA肿瘤数据库的多维数据积累;●本研究试图从信号通路角度对TCGA数据库中实体瘤样本进行多维数据整合分析,以系统评估不同肿瘤亚型在典型信号通路中的变异基因特征、共发生与互斥关系,从而发掘潜在联合靶向药物方案。
摘要方法:通过对TCGA数据库中涉及33种肿瘤类型,64个肿瘤亚型,共9125个肿瘤样本的体细胞突变、拷贝数变异、mRNA表达、基因融合和基因组甲基化数据,基于10个典型肿瘤相关信号通路,进行统一的标准数据处理与多维度整合分析。
结果:(1)多维整合数据分析得到10个典型肿瘤相关信号通路的变异图谱;(2)鉴定出10个典型信号通路中的driver基因集;(3)发现57%的肿瘤样本于这些信号通路中包含至少1个潜在actionable变异,同时,89%样本至少包含1个driver 变异,30%样本包含多个可靶向变异;(4)鉴定了通路中变异的共显与互斥表现模式。
结论:首次针对TCGA数据库进行大规模的肿瘤相关信号通路多维数据整合分析,为肿瘤信号通路大数据挖掘提供了一种统一的标准化数据处理流程和分析框架;鉴定了经典信号通路中的变异模式,actionable变异的共显与互斥模式提示潜在的联合治疗可能。
数据样本与癌种分布●共包含TCGA数据库中9125个肿瘤样本,涉及33个癌种,64个亚型;●左图显示各肿瘤类型与亚型的数量分布与占比。
(最内圈不同颜色标明肿瘤组织部位,次内圈不同颜色表示不同肿瘤类型,最外圈不同颜色深浅代表不同肿瘤亚型)数据处理与分析流程●首先通过标准数据处理、pathway数据库、文献报道pathway、TCGA已发表相关pathway文献结果等来源,初步确定待选肿瘤相关pathways;●其次通过各种已知基因功能数据库定义driver变异,并通过人工筛选与校验待选信号通路;●最后对筛选出的典型pathway进行后续相关整合分析与数据展示(仅对信号通路中已知driver变异和统计学显著的基因进行分析)。
TCGA的28篇教程-对TCGA数据库的任意癌症中任意基因做生存分析本教程目录:•首先使用cgdsr获取表达数据集临床信息•临床资料解读•简单的KM生存分析•有分类的KM生存分析•根据基因表达量对样本进行分组做生存分析•cox生存分析•某基因突变与否也可以用来分组•基因的拷贝数也可以进行分组•批量进行分组并且做生存分析生存分析一般来说是针对RNA表达数据,可以说mRNA-seq的转录组数据,也可以说miRNA-seq数据,或者基因表达芯片的表达量值。
生存分析,大多就是说的KM方法估计生存函数,并且画出生存曲线,然后还可以根据分组检验一下它们的生存曲线是否有显著的差异。
在R中,有个包survival做生存分析就很方便!只需要记住和熟练使用三个函数:•Surv:用于创建生存数据对象•survfit:创建KM生存曲线或是Cox调整生存曲线•survdiff:用于不同组的统计检验首先使用cgdsr获取表达数据集临床信息既然是要说明如何对任意癌症的任意基因做生存分析,那么我们首先需要理解cgdsr下载TCGA任意数据的用法(见之前的教程),下面的例子是获取TCGA数据库的乳腺癌的BRCA1和BRCA2基因的表达,以及涉及到的病人的临床资料。
rm(list = ls())library(cgdsr)library(DT)mycgds <- CGDS("/public-portal/")mycancerstudy = 'brca_tcga'## 下面的代码可以不需要运行,因为已经保存好了用来做生存分析的数据。
### 但是需要看懂代码,这样才能做任意癌症的任意基因的任意数据的生存分析;if(F){getCaseLists(mycgds,mycancerstudy)[,1]getGeneticProfiles(mycgds,mycancerstudy)[,1]mycaselist ='brca_tcga_rna_seq_v2_mrna'mygeneticprofile = 'brca_tcga_rna_seq_v2_mrna'choose_genes=c('BRCA1','BRCA2')## get expression dataexpr=getProfileData(mycgds,choose_genes,mygeneticprofile,mycaselist)## get mutation datamut_df <- getProfileData(mycgds,caseList ="brca_tcga_sequenced",geneticProfile = "brca_tcga_mutations",genes = choose_genes)mut_df <- apply(mut_df,2,as.factor)mut_df[mut_df == "NaN"] = ""mut_df[is.na(mut_df)] = ""mut_df[mut_df != ''] = "MUT"## get copy number datacna <- getProfileData(mycgds,caseList ="brca_tcga_sequenced",geneticProfile = "brca_tcga_gistic",genes = choose_genes)rn=rownames(cna)cna <- apply(cna,2,function(x)as.character(factor(x, levels = c(-2:2),labels = c("HOMDEL", "HETLOSS", "DIPLOID", "GAIN", "AMP "))))cna[is.na(cna)] = ""cna[cna == 'DIPLOID'] = ""rownames(cna)=rn# Get clinical data for the case listmyclinicaldata = getClinicalData(mycgds,mycaselist)save(expr,myclinicaldata,cna,mut_df,file='survival_input.Rdata')}load(file='survival_input.Rdata')DT::datatable(expr)上述代码取决于网速,我已经下载整理好了:survival_input.Rdata 数据,避免每次重复这个教程重新下载的尴尬DT::datatable(myclinicaldata,extensions = 'FixedColumns',options = list(#dom = 't',scrollX = TRUE,fixedColumns = TRUE))## Warning in instance$preRenderHook(instance): It seems your data is too## big for client-side DataTables. You may consider server-side processing:## http://rstudio.github.io/DT/server.html可以看到所谓的表达矩阵就是每个基因在各个样本的表达量,只不过是要注意单位,可以是RPKM,TPM等。
TCGA数据库生存分析正文简介生存分析(Survival analysis)是指根据试验或调查得到的数据对生物或人的生存时间进行分析和推断,研究生存时间和结局与众多影响因素间关系及其程度大小的方法,也称生存率分析或存活率分析。
生存分析适合于处理时间-事件数据,生存时间(survival time)是指从某起点事件开始到被观测对象出现终点事件所经历的时间,如从疾病的“确诊”到“死亡”。
类型生存时间有两种类型:完全数据(complete data)指被观测对象从观察起点到出现终点事件所经历的时间;截尾数据(consored data)或删失数据,指在出现终点事件前,被观测对象的观测过程终止了。
由于被观测对象所提供的信息是不完全的,只知道他们的生存事件超过了截尾时间。
截尾主要由于失访、退出和终止产生。
生存分析方法大体上可分为三类:非参数法、半参数方法和参数法,用Kaplan-Meier曲线(也称乘积极限法Product limit method)和寿命表法(Life table method)估计生存率和中位生存时间等是非参数的方法,半参数方法指Cox比例风险模型,参数方法指指数模型、Weibull模型、Gompertz模型等分析方法。
非参数法寿命表时描述一段时间内生存状况、终点事件和生存概率的表格,需计算累积生存概率即每一步生存概率的乘积,可完成对病例随访资料在任意指定时点的生存状况评价。
survival包中包括了所有生存分析所必须的函数,生存分析主要是把数据放入Surv object,通过Surv()函数做进一步分析。
Surv object是将时间和生存状况的信息合并在一个简单的对象内,Surv(time, time2, event,type=c(‘right’, ‘left’, ‘interval’, ‘counting’, ‘interval2’, ‘mstate’),origin=0),time为生存时间,time2为区间删失的结束时间,event为生存状况,生存状况变量必须是数值或者逻辑型的。
数据挖掘TCGATCGA是什么?由美国05年发起的癌症和肿瘤基因图谱(TCGA)计划,旨在应用基因组分析技术研究癌症中的基因组变化,做了大规模的基因组测序,样本量过万,包含了三十多种癌症,其中尤其宝贵的是这些样本都有很详细的预后随访信息,08年出了第一篇文章,之后陆陆续续各种大文章出来。
数据及类型汇总数据及类型汇总TCGA包含了哪些数据?1、临床样本信息:Biospecimen、Clinical2、测序数据:主要采用了RNA-Seq、WXS、miRNA-Seq、GenotypingArray、Methylation Array这五种方式对样本进行测序。
对测序数据按照一定的分析程度进行分层,共分为四层:level1、level2、level3、level4,按照不同的层次的数据提供给科研人员使用,level3、level4的数据一般都开放下载的,level1是最原始的数据,level2是做了进一步的处理的这些数据一般是不开放的,需要申请才能下载(申请也挺难)。
TCGA中的RNA-Seq数据顾名思义就是转录组测序·TCGA上的转录组数据采用的是全转录组测序,其中包含了各种非编码RNA,所以一般下载的RNA-Seq数据中包含了lncRNA、mRNA、假基因等等;·目前可以公开下载的是RNA-Seq定量表达数据,主要三种形式:HT-Seq-FPKM,HT-Seq-UQ-FPKM,HT-Seq-Counts;TCGA用的gff文件是gencode.v22.annotation.gtf名词解释·FPKM:用来衡量转录本表达丰度的一种量度方式;·Counts:测序的reads中比对到某个基因上的计数;·UQ-FPKM:通过上四分位点进行标准化后的FPKM;·gff文件:用来描述基因组上各种基因、转录本等信息的文件。
TCGA中的miRNA-Seq数据·miRNA:miRNA是一类由内源基因编码的长度约为22 个核苷酸的非编码单链RNA分子,生物中非常重要的一类非编码小RNA,其在生物体的调控中具有非常重要的作用,在人中大约三分之一的基因受到miRNA的调控;·TCGA提供了miRNA-Seq的测序数据结果,采用的数据库背景为miRBase v21;·目前公开提供下载的数据主要有两种:miRNAExpression Quantification、Isoform Expression Quantification,其中Isoform Expression Quantification数据中包含了成熟体miRNA;·分别提供了Counts和FPKM格式的定量数据。
第 6 卷 第 6 期2020 年 12 月生物化工Biological Chemical EngineeringVol.6 No.6Dec. 2020基于TCGA数据库构建肝癌Ten-miRNAs风险评估模型及预后分析陈俊光(石河子大学 生命科学学院,新疆石河子 832003)摘 要:目的:寻找可作为肝癌生物标记物的miRNAs,构建肝癌风险评估模型。
方法:利用TCGA数据库的肝癌患者高通量测序数据和临床数据集进行肿瘤组织和正常组织之间miRNAs的差异分析。
使用Cox单因素回归分析评估和不良预后相关的miRNAs,筛选差异表达中上调的miRNAs进行Cox多因素回归分析,建立风险评估模型。
结果:与周围正常组织差异表达的miRNAs有247个,其中228个上调,19个下调;进一步分析显示,有23个miRNAs的过表达和不良预后相关(P<0.05),从中筛选出10个miRNAs作为预测肝癌不良预后的生物标志物组合。
结论:Ten-miRNAs特征模型在预测肝癌患者存活风险方面具有良好的灵敏度和特异性。
关键词:肝癌;TCGA;差异表达;Cox回归分析;风险评估中图分类号:R730.7 文献标识码:AA Ten-miRNAs Expression Signature PRSS and Prognosis Analysis for Liver Hepatocellular Carcinoma were Established Based on TCGA DatabaseCHEN Junguang(College of Life Sciences, Shihezi University, Xinjiang Shihezi 832003)Abstract: Objective: In order to find miRNAs that can be used as biomarkers for liver cancer, a prognostic risk score system (PRSS) for Liver hepatocellular carcinoma (LIHC). Methods: Download High-throughput Sequencing data and clinical data sets of patients in the TCGA database to analyze the differential expression in miRNAs between tumor and normal tissues. Cox univariate regression analysis was used to evaluate miRNAs related to poor prognosis, and miRNAs that were up-regulated in differential expression were screened for Cox multivariate regression analysis to establish a PRSS. Results: There were 247 miRNAs differentially expressed from surrounding normal hepatic tissues, of which 228 were up-regulated and 19 were down-regulated. Further analysis showed that overexpression of 23 miRNAs was associated with poor prognosis (P<0.05), and 10 miRNAs were selected as biomarkers for predicting poor prognosis of LIHC. Conclusions: Ten-miRNAs-PRSS has good sensitivity and specificity in predicting the survival risk of LIHC, but the specific role needs further experimental analysis.Keywords: LIHC; TCGA; differential expression of gene; cox regression analysis; PRSS肝癌(Liver hepatocellular carcinoma,LIHC)是指发生于肝脏的原发性或者继发性肿瘤。
《基于TCGA数据库乳腺癌IncRNA的分析研究》一、引言乳腺癌是全球女性最常见的恶性肿瘤之一,其发病率逐年上升,对女性健康构成严重威胁。
随着生物信息学和基因组学的发展,越来越多的研究开始关注非编码RNA(ncRNA)在疾病发生、发展中的作用。
其中,长链非编码RNA(IncRNA)因其特殊的调控作用和复杂的生物学功能,成为研究的热点。
TCGA(The Cancer Genome Atlas)数据库作为全球最大的癌症基因组数据库之一,为乳腺癌IncRNA的研究提供了丰富的数据资源。
本文旨在基于TCGA数据库,对乳腺癌IncRNA进行深入分析研究,以期为乳腺癌的预防、诊断和治疗提供新的思路和方法。
二、材料与方法1. 数据来源本研究采用的数据来自TCGA数据库中的乳腺癌相关数据,包括基因表达谱、临床信息等。
2. 研究方法(1)数据预处理:对基因表达谱数据进行质量评估和预处理,去除低质量和异常值数据。
(2)IncRNA筛选:基于基因表达谱数据,筛选出在乳腺癌组织中显著差异表达的IncRNA。
(3)功能分析:通过生物信息学分析方法,对筛选出的IncRNA进行功能分析,包括基因共表达网络分析、基因集富集分析等。
(4)验证实验:结合临床样本,对筛选出的关键IncRNA进行实时荧光定量PCR验证。
三、结果与分析1. 差异表达IncRNA的筛选结果通过数据分析,我们筛选出在乳腺癌组织中显著差异表达的IncRNA共计XX个,其中XX个为上调表达,XX个为下调表达。
这些IncRNA在乳腺癌的发生、发展过程中可能发挥重要的调控作用。
2. 功能分析结果通过对筛选出的IncRNA进行功能分析,我们发现这些IncRNA主要参与细胞增殖、凋亡、侵袭和转移等生物学过程。
其中,某些关键IncRNA与乳腺癌的预后密切相关,可能成为乳腺癌诊断和治疗的潜在靶点。
3. 实时荧光定量PCR验证结果为了进一步验证筛选出的关键IncRNA的准确性,我们结合临床样本进行了实时荧光定量PCR验证。
TCGA数据库在线使⽤本⽂包括了TCGA本站中数据的浏览、下载,尤其是TCGA改版后的功能介绍(增加了OncoGrid展⽰),然后是cBioPortal,TCGA数据在线提供的分析类型最多的⼀个平台,再是FIREBROWSE,⽐较不错的在线展⽰和⽅便的数据下载功能。
TCGA主站TCGA分析了11,000个病⼈的33种肿瘤的7个不同层⾯的数据,共获得2.5 PB数据。
意在解析癌症发⽣的分⼦接触、肿瘤的亚型和治疗靶点等。
TCGA⽹站主要提供的是数据的浏览和下载功能,可以根据项⽬、个体、数据类型、肿瘤类型等筛选需要的数据,使⽤TCGA提供的⼯具下载,进⼀步分析。
TCGA项⽬促成了不少的⾼⽔平⽂章,对这些⽂章的阅读是对癌症知识的学习,也可以很好的扩展研究思路。
如果你需要帮助,WIKI是最好的伙伴。
最新版的TCGA 增加了⼀些分析的功能,主要是展⽰基因的信息、突变频率、突变位点分布、OncoGrid信息等。
在搜索框搜索基因,癌症类型,个体编号会有不同的结果体验。
查看基因在哪种癌症中突变最频繁突变位点在基因和功能域的分布,纵轴表⽰突变个体数⽬。
基因每个位点的突变频率,为上图纵轴信息的表格展⽰。
乳腺癌中突变频率最⾼的基因和病⼈⽣存曲线500个突变最多的个体和50个最⾼突变的基因,顶部柱状图代表每个个体中这50个基因的突变位点数⽬,右侧柱状图表⽰含有每个基因突变位点的个体数⽬,热图不同颜⾊代表不同的突变类型,下⽅2个颜⾊条代表临床信息和数据类型,右侧的第⼀个颜⾊条代表该基因是否是Cancer Gene Census (The Cancer Gene Census is a list of genes with substantial published evidence in Oncology.),第⼆个颜⾊条代表突变影响到的个体数。
cBioPortal功能强⼤的TCGA再分析平台cBioPortal可查询选定的癌症中某⼀通路的基因或⽤户⾃定义的多个基因的信息,多个基因的结果部分合并展⽰,部分独⽴展⽰。