201208-欧盟EMEA关于变更的要求
- 格式:ppt
- 大小:154.00 KB
- 文档页数:22

2003年10月欧盟药品评价管理局(EMEA)起草了直接接触塑料包装材料指导原则(GUIDELINE ON PLASTICIMMEDIATE PACKAGING MATERIALS),并与2005年12月1日发布.该指导原则根据风险级别,对于直接接触原料药或制剂的塑料包材应进行哪些研究,如何在申报资料中呈现,提供了指导意见。
这一指导原则对于我国直接接触药品的塑料包材研究具有很高的借鉴意义。
因此笔者进行了翻译,特此供业界参考研究。
以下为指导原则正文。
目录1 介绍1。
1 目标1。
2 概述1.3 一般原则2 在申请上市文件中的位置3 应提交的数据3。
1 总体信息3.2 质量标准4 提取研究5 相互作用研究5。
1 迁移(浸出)研究5。
2 吸附研究6 毒理学资料/文献7 术语解释附件1 申报资料决策树附件2 塑料包装材料申报资料决策树附件3 提交信息对照表1 介绍1.1 目标制定本指导原则旨在替代《医药产品管理办法》3AQ10a的“直接接触塑料包装材料指导原则”,同时进一步强调在原料药和制剂申请上市时,应针对其直接接触药品的塑料包装材料提供相关信息。
本指导原则涉及人用药品和兽药所用的直接接触药品的塑料包装材料的申请.对于人用药品,本指导原则涉及欧盟法规2003/63/EC(法规2001/83/EC的修正版)附录I第一部分第3单元的章节3.2。
1.6、3。
2。
2.2和3.2。
2.7;对于兽药,则涉及欧盟法规2001/82/EC的附录I第二部分的章节A、C和G。
1.2 概述本指导原则囊括了对直接接触药品塑料包装材料的具体要求。
对于其他包装材料或容器密封系统的特性,如包材性能,本指导原则不会考虑为它们制定一个合适的总体要求。
本指导原则范围仅限于直接接触药品塑料包装材料,也就是与原料药或制剂发生直接接触的包装材料,它们可能只是容器密封系统中的容器、封盖或其他部件的某一部分。
弹性体、天然和人工橡胶不在本指导原则范围之内.本指导原则不适用于对采用已批准包材的上市药品进行回顾性研究。

变更管理需要遵循的法规要求一、《企业安全生产标准化基本规范》(GB/T33000-2016)5.5.1.4变更管理企业应制定变更管理制度。
变更前应对变更过程及变更后可能产生的安全风险进行分析,制定控制措施,履行审批及验收程序,并告知和培训相关从业人员。
二、《危险化学品从业单位安全标准化通用规范》AQ3013-20086.6.5变更7.6.5.1企业应严格执行变更管理制度,履行下列变更程序:1)变更申请:按要求填写变更申请表,由专人进行管理;2)变更审批:变更申请表应逐级上报主管部门,并按管理权限报主管领导审批;3)变更实施:变更批准后,由主管部门负责实施。
不经过审查和批准,任何临时性的变更都不得超过原批准范围和期限;4)变更验收:变更实施结束后,变更主管部门应对变更的实施情况进行验收,形成报告,并及时将变更结果通知相关部门和有关人员。
5.6.5.2企业应对变更过程产生的风险进行分析和控制。
三、《关于加强化工过程安全管理的指导意见》(二十二)建立变更管理制度。
企业在工艺、设备、仪表、电气、公用工程、备件、材料、化学品、生产组织方式和人员等方面发生的所有变化,都要纳入变更管理。
变更管理制度至少包含以下内容:变更的事项、起始时间,变更的技术基础、可能带来的安全风险,消除和控制安全风险的措施,是否修改操作规程,变更审批权限,变更实施后的安全验收等。
实施变更前,企业要组织专业人员进行检查,确保变更具备安全条件;明确受变更影响的本企业人员和承包商作业人员,并对其进行相应的培训。
变更完成后,企业要及时更新相应的安全生产信息,建立变更管理档案。
(二十三)严格变更管理。
工艺技术变更。
主要包括生产能力,原辅材料(包括助剂、添加剂、催化剂等)和介质(包括成分比例的变化),工艺路线、流程及操作条件,工艺操作规程或操作方法,工艺控制参数,仪表控制系统(包括安全报警和联锁整定值的改变),水、电、汽、风等公用工程方面的改变等。

eugmp附录1变更解读关于eugmp附录1变更解读的文章。
注:在文章中,"eugmp"指欧洲药典通用制药法规(European Union Good Manufacturing Practice,以下简称"EU-GMP"),"附录1"是指EU-GMP中的附录1,该附录关于药品生产的基本原则和指南。
第一步:了解eugmp附录1的基本内容在撰写关于eugmp附录1变更解读的文章之前,我们首先需要了解eugmp附录1的基本内容。
eugmp附录1提供了欧盟对药品生产质量管理系统的要求,包括建筑物和设备规范、人员培训和责任、文件管理和记录、原材料采购和控制、生产工艺控制、环境监测等方面的指导原则。
第二步:探讨eugmp附录1变更的原因接下来,我们需要探讨eugmp附录1发生变更的原因。
在过去的几十年里,药品生产和监管环境发生了巨大的变化,包括技术进步、新药品开发和国际贸易的增加等。
由于这些变化,欧盟药品监管机构认为有必要对eugmp附录1进行修订,以确保药品制造商能够满足新的质量和安全要求。
第三步:介绍eugmp附录1的具体变更内容现在,我们可以介绍eugmp附录1的具体变更内容。
例如,变更可能涉及对环境监测要求的修改,包括空气和水质的监测,以及对环境控制系统的要求。
另外,变更还可能包括对设备验证和清洁验证的更严格要求,以及对原材料供应链和分销网络的更加严格的监控。
第四步:分析eugmp附录1变更对药品制造商的影响下一步,我们需要分析eugmp附录1变更对药品制造商的影响。
新的要求可能需要制造商进行改进、投资和培训,以确保其符合新的规定。
这可能会导致生产成本的增加,但同时也能提高药品的质量和安全性,增强欧盟市场对这些产品的信任。
第五步:讨论制药厂如何应对eugmp附录1的变更最后,我们需要讨论制药厂如何应对eugmp附录1的变更。
制药厂可以采取一系列措施来满足新的要求,例如开展内部分析和评估,建立新的程序和培训计划,与供应商和分销商进行合作,以确保整个供应链都符合新的要求。


4M1实施管理办法1 目的为保证供货质量的稳定和一致性,供应商4M1E(4M1E 即人( Man )、设备( Machine )、材料(Material) 、作业方法(Method) 、环境( Environment ))变更时需要申报,相关变更需在受控状态下进行。
所谓4M1E 变更,是指与供应商的制造工序相关的条件(部件规格、材料、检查方法、合作公司、生产场所、作业方法、制造方法、制造条件、夹具、生产设备、模具、作业人员等)发生变化的情况。
2 适用范围所有我司合格供应商。
3 变更中的质量保证为了保证零部件的质量稳定,供应商应确定并执行针对4M1E 变更的要求,明确变更管理内容,进行质量验证,切实执行本平台所述的内容。
4 变更管理的区分有关变更的内容及其管理区分,可参阅附表1 “关于变更内容的区分”。
管理区分有如下两类:自我管理、变更申请。
供应商应根据实际情况进行变更的区分与管理,如对与变更申请相关的变更区分产生疑义时,可向我司技术质量部相关人员核实。
自我管理“自我管理区分”即附表1中“自我管理”的变更。
这种情况下,不必向我司提出申报。
供应商应调整公司体制,实行自我管理。
变更申请“变更申请”表示附表1 中属于“申请”的变更。
“变更申请书”请参阅附件3。
供应商应在申请书上注明变更内容、变更原因、验证结论等事项,由本公司相关主管部门(质量部门)审批盖章并签名后,提交给我司经营部。
在提交变更申请书时,供应商应提交详细的验证资料以证明变更的合理性。
在收到我司书面同意变更的审核确认后,供应商与我司经营部、技术质量部就供货日期达成协议,方可交纳批次变更品。
并对变更后的产品进行标识和单独报检,并注明。
变更步骤可参阅附图 2 “供应商4M1E变更申请信息处理流程”。
对于附表 1 中未涉及到的变更,如果也会影响到产品的质量,供应商应根据实际造成的影响来决定是否申报;对于有害物质有影响的4M1E变更,供应商要在定期提供ROHS宣告表的基础上,及时提交变更申请书。

变更管理的法规要求
变更管理的法规要求是根据不同国家和地区的监管机构和行业标准而有所不同。
然而,下面列出的是一些常见的变更管理法规要求:
1. 风险评估和管理:变更管理过程应包括对变更带来的潜在风险进行评估和管理。
这可能包括评估对安全、法规合规性、业务连续性等方面的风险。
2. 批准和授权:变更应该经过适当等级的批准和授权。
这意味着有人应对变更提出请求进行评估,并有权决定是否批准变更。
3. 记录和审核:变更管理过程需要有详细的记录,包括变更请求、批准、实施和验证的信息。
这些记录可以用于审计和事后评估。
4. 变更控制板或委员会:某些行业可能要求设立独立的变更控制板或委员会,负责评审和决定变更请求。
这个委员会应由相关部门的代表组成。
5. 测试和验证:变更管理要求对变更进行严格的测试和验证。
这可以包括在一个非生产环境中进行试验和模拟,以确保变更对系统和业务的影响可以被准确评估。
6. 交流和沟通:变更管理要求在变更实施之前与相关利益相关者进行充分的沟通和协商。
这可以包括向利益相关者提供变更计划、风险评估和变更的预期影响等信息。
需要注意的是,这些要求可能只是变更管理法规的一部分。
具体的要求可能因行业、国家或地区的特殊情况而有所不同。
因此,在实施变更管理时,组织应根据适用的法规和标准来制定和执行相关的程序和政策。

欧洲议会与欧盟理事会关于转基因生物的可追溯性和标识及由转基因生物制成的食品和饲料产品的可追溯性,并对指令2001/18/EC进行修改的第1830/2003号条例2003年9月22日经下列指令修订:序号页码日期L 311 1 2008年11月21日2008年10月22日欧洲议会与欧盟理事会第1137/2008号条例(EC)欧洲议会与欧盟理事会考虑到欧洲共同体缔结的《条约》以及其中第95(1)条,考虑到欧盟委员会建议书,考虑到欧洲经济与社会委员会的意见,考虑到区域委员会的意见,根据《条约》第251条款中规定的程序,作出决定:鉴于:(1) 欧洲议会和理事会于2001年3月12日通过的有关有意向环境排放转基因生物(GMOs)的2001/18/EC指令要求成员国采取措施来确保经授权的GMOs在投入市场的所有阶段具有可追溯性和标识性。
(2) 国家法律、条例、管理规定(关于GMOs产品或包含GMOs产品的可追溯性和标识以及由GMOs制成的食品和饲料产品的可追溯性)之间存在的不同可能会阻碍这些产品的自由交易,将导致不平等和不公平竞争。
为GMOs的可追溯性和标识创立协调的欧洲共同体框架将有助于国内市场的有效运转。
因此,对指令2001/18/EC进行修改。
(3) GMOs的可追溯性要求应简化以下过程:召回对人体健康、动物健康、生态环境(包括生态系统)产生不可预见的不良影响的产品,监测检查其影响,尤其是对环境的影响。
按照预防原则,可追溯性也可简化风险管理措施的实施。
(4) 根据欧洲议会和理事会2003年9月22日条例(EC)1829/2003对转基因食品和饲料的要求,由GMOs制成的食品和饲料产品应具有可追溯性,以此简化此产品的准确标识来确保经营者和客户可以获得准确信息,使他们可以通过有效方式自由选择,并且可以对标识索赔进行控制和检验。
对由GMOs制成的食品和饲料产品提出的要求应一致来避免产品最终用途改变造成信息不连续。

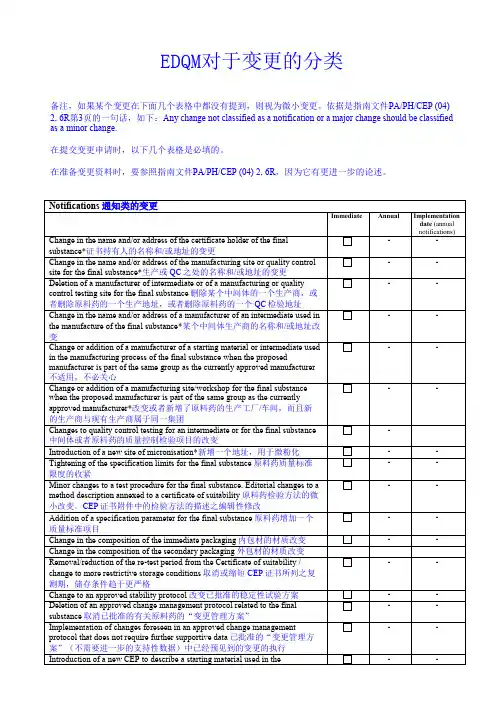
Date of implementation: 1 March 2010Introduction:The holder of a Certificate of suitability shall inform the EDQM of any change to the information in the certification dossier by sending an application form and all necessary documents demonstrating that the conditions laid down in the present guideline are met.Classification of changesThe changes have been classified in three categories (notification/minor/major) depending on the potential impact of the change on the quality of the final substance. These three categories are based on those (IA-IAIN/IB/II) of the Commission Regulation (EC) No 1234/2008 concerning the examination of variations to the terms of marketing authorisation for medicinal products for human use and veterinary medicinal products.Any change not classified as a notification or a major change should be classified as a minor change except in the following cases where a new application should be submitted:- addition of a new route of synthesis and/or a new manufacturing site where the specifications of the final substance are different from the one already approved- transfer to a new holder that is not the same legal entity as the approved one, where the transfer does not occur because of a merger or because the company is sold, and where the manufacturer does not take out the Certificate of suitability in their own name.The changes related to Ph. Eur. monograph revisions or any other regulatory requirements are treated separately and generally initiated by the EDQM.执行日期:2010年3月1日介绍:欧洲药典适用性证书持有人必须向EDQM报告所有与申报文件有关的变更,申报时应填写申请表格和所有必要的资料,证明变更符合现行指南的规定。

材料变更报告随着科技的不断发展和市场的不断变化,材料变更成为制造业中不断面临的问题。
材料变更通常是指在产品生产过程中,更改了产品所用的材料的种类、配比或品牌等,这不仅可能影响产品的性能、品质和安全性,还可能影响消费者的购买决策和评价。
因此,制造商需要在材料变更前向相关机构提交材料变更报告,以确保产品的质量和安全性,并获得优良的市场口碑。
一、美国食品药品监督管理局(US FDA)关于材料变更报告的要求美国食品药品监督管理局要求制造商必须在进行材料变更之前提交变更报告,并获得审批。
其中,要求变更的材料不能影响产品的基本特性、安全性和效用;另外,还必须提供详细的变更控制程序、质量保证体系和效果验证程序。
二、欧洲航空安全局(EASA)关于材料变更报告的要求欧洲航空安全局要求制造商必须提交材料变更报告,并获得批准。
其中,要求变更的材料必须满足产品的技术规范和标准,同时必须提供详细的测试数据和材料使用手册等资料。
三、国家食品药品监管总局关于材料变更报告的要求国家食品药品监管总局要求制造商必须在进行材料变更之前提交变更报告,并获得批准。
其中,要求变更的材料必须符合我国相关的法律法规和标准,同时必须提供详细的安全性评估和效果验证等数据和资料。
以上三个案例表明,材料变更报告对于确保产品质量和安全性至关重要。
制造商必须认真遵守相关机构的要求和规定,确保变更的材料符合产品技术规范和标准,并提供详细的数据和资料,以便相关机构及时审批并公开公示。
只有这样,制造商才能获得消费者的信任和认可,赢得市场的竞争优势。
此外,材料变更报告也是制造商对产品质量和安全性负责任的表现。
在材料变更发生后,制造商需要对变更的影响进行评估和验证,并及时向市场和消费者公开披露,以保持透明度和诚信度,避免因变更而带来的负面影响。
当然,制造商在提交材料变更报告时也需要注意一些问题。
比如,在说明变更的原因时,不应该有虚假宣传和误导消费者的行为;在提交测试数据和效果验证资料时,应该确保数据的真实有效,并且符合相关的标准和试验方法。

欧盟国会与市政委员会第1223/2009法规2009.11.30化妆品(重新制定)(随带相关电子索引)鉴于: (3)第一章 (9)条款1 (9)条款2 (9)第二章 (10)条款3 (10)条款4 (11)条款5 (11)条款6 (12)条款7 (12)条款8 (13)条款9 (13)第三章 (13)条款10 (13)条款11 (14)条款12 (14)条款13 (15)第四章 (16)条款14 (16)条款15 (17)条款16 (18)条款17 (20)第五章 (20)条款18 (20)第六章 (21)条款19 (21)条款20 (23)条款21 (24)第七章 (24)条款22 (24)条款23 (24)条款24 (25)第八章 (25)条款25 (25)条款26 (27)条款27 (27)条款28 (27)第九章 (28)条款29 (28)条款30 (28)第十章 (28)条款31 (28)条款32 (29)条款33 (29)条款34 (29)条款35 (30)条款36 (30)条款37 (30)条款38 (30)条款39 (31)条款40 (31)附录I (32)A部分——化妆品安全信息 (32)B部分——化妆品安全评估 (34)附录II到VI的前言 (35)附录II (36)附录III (36)附录IV (36)附录V (36)附录VI (36)附录VII (37)附录VIII (38)附录IX (38)A部分 (38)B部分 (38)附录X (38)欧盟国会与市政委员会,鉴于欧洲成员国建立的条约和其95号文件,鉴于委员会的提议,鉴于欧洲经济和社会团体的观点,依据程序必须法令写于条约第251号文件,鉴于:1. 1976年7月27日成员国颁布的关于化妆品的法规指令76/768/EEC已经过几次重大修订。
因有更多需修订的地方,为了指令更清晰,在这特定情况下此法规需要以单独文件形式重新制定。
医疗器械技术变更法规一、概述在医疗器械领域中,技术变更是指对已经上市的医疗器械进行修改或改进,以提高其性能、功能或质量,或适应新的治疗方法和技术要求。
技术变更的合规性与推广和使用这些器械的安全性、有效性密切相关。
因此,各国纷纷制定了一系列法规和规范来调整医疗器械技术变更的管理。
二、国际法规与标准1. 国际电工委员会(IEC)IEC制定了一系列标准,包括技术规范和测试方法,以指导医疗器械技术变更的研发、评估和控制。
这些标准为各国建立国内法规提供了重要的参考依据。
2. 美国FDA法规美国食品药品监督管理局(FDA)针对医疗器械技术变更制定了较为严格的管理要求。
根据FDA的规定,技术变更分为三个类别:重大、非重大和适度。
不同类别的技术变更需要符合不同的要求和程序。
3. 欧盟医疗器械技术变更指南欧盟对医疗器械技术变更的管理采取专门的指南,并制定了特定的程序和要求。
根据欧盟指南,技术变更应根据器械的风险等级进行分类,并采取相应的管理措施。
三、国内法规与标准1. 中国医疗器械监察标准与规范中国国家药品监督管理局(NMPA)制定了一系列医疗器械技术变更的监察标准与规范。
这些标准与规范包括了技术变更的分类、风险评估、文件要求等内容,为企业在国内市场推广医疗器械提供了指导。
2. 专利法与知识产权保护在技术变更领域,专利法和知识产权保护扮演着重要角色。
企业在进行技术变更时,需要注意是否侵犯了他人的专利权,以避免可能的法律纠纷。
四、医疗器械技术变更管理流程1. 技术变更申请与评估企业向相关监管机构提出技术变更申请,同时提交变更申请文件和相关测试报告。
监管机构对申请进行评估,以确定技术变更是否符合相关法规和标准的要求。
2. 风险评估与验证根据风险管理原则,对技术变更进行风险评估,并进行必要的验证实验。
风险评估结果应根据风险等级进行分类,并采取相应的管理措施。
3. 文件管理与备案企业应建立技术变更的文件管理制度,并将申请文件、评估报告、验证实验结果等相关文件进行归档备案,以便后续监督和审计。
欧盟修改有关食品添加剂法令
佚名
【期刊名称】《中国饲料添加剂》
【年(卷),期】2006(000)007
【摘要】欧洲委员会计划修改有关食品添加剂和增甜剂的欧盟法令。
所推荐的修改文献包括肉类产品中严格使用亚硝酸盐和硝酸钾,同时与法庭有关规定和欧洲食品安全局有关这些物质含量水平安全而不会对肉类产品造成危害的意见是一致的。
此建议也允许使用7种新的食品添加剂及已经允许使用的其他某些添加剂。
【总页数】2页(P51-52)
【正文语种】中文
【中图分类】TS202
【相关文献】
1.欧盟修改有关含铝食品添加剂使用条件和标准
2.欧盟修改含铝食品添加剂使用条件和标准
3.欧盟修改含铝食品添加剂使用条件和标准
4.欧盟修改含铝食品添加剂使用条件及标准
5.欧盟新传媒法对于置入式广告与广告法令的修改
因版权原因,仅展示原文概要,查看原文内容请购买。
药物安全性监察——EMEA要求从市场撤出Acomplia
张宇(摘)
【期刊名称】《国外药讯》
【年(卷),期】2009(000)001
【摘要】欧洲医药管理局(EMEA)要求暂时从市场撤出Sanofi-Aventis公司的
肥胖治疗药Acomplia(rimonabant)(Ⅰ)。
EMEA人用药委员会(CHMP)
认为,这种大麻素类受体阻滞剂的利益不再超过它的风险。
该药于2006年6月在欧盟国家获得批准,作为有相关危险因素的肥胖或超重病人饮食控制和运动的辅助手段。
【总页数】0页(P35)
【作者】张宇(摘)
【作者单位】无
【正文语种】中文
【中图分类】R95
【相关文献】
1.药物安全性监察——锶和Prexige被列入EMEA安全行动名单 [J], 金伟秋(摘)
2.FDA要求未经批准的曲美苄胺撤出市场 [J], 曹菊(摘)
3.药物安全性监察:GAO要求改善FDA的售后决策 [J], 杨绍杰(摘)
4.药物安全性监察:09082 Bayer自动从美国撤出Baycol [J], 陈贞
5.美FDA要求丙氧芬撤出美国市场 [J], 马培奇
因版权原因,仅展示原文概要,查看原文内容请购买。
欧盟就上市产品变更征求意见
无
【期刊名称】《国外药讯》
【年(卷),期】2009(000)007
【摘要】欧盟委员会公开征求有关新变更管理规定指南草案的意见。
委员会于3月份曾征求有关新变更管理规定实施的指南草案的意见。
【总页数】1页(P3)
【作者】无
【作者单位】无
【正文语种】中文
【中图分类】R95
【相关文献】
1.A股上市公司股票简称变更效应研究——基于2015年变更股票简称的A股上市公司数据统计 [J], 林晓梦
2.上市公司审计师变更特征、行业自律管理与审计质量研究——基于上市公司年报事务所变更报备信息研究 [J], 汪月祥;王喆祥;孙娜
3.产品市场竞争、CEO变更与现金股利支付——来自我国A股上市公司的经验证据 [J], 吕沙
4.商务部关于《外商投资企业设立及变更备案管理办法(征求意见稿)》公开征求意见的通知 [J],
5.诗华马立克产品上市发布会在南宁举行标志着华都诗华第一个符合欧盟标准的产品正式上市 [J], 本刊讯
因版权原因,仅展示原文概要,查看原文内容请购买。