色谱分析(中国药科大学)超高效液相色谱(UPLC)
- 格式:doc
- 大小:315.50 KB
- 文档页数:9


超高效液相色谱(UPLC)超高效液相色谱技术(ultra performance liquid chcromatography,简称UPLC)是一种综合了小颗粒填料、非常低系统体积(死体积)及快速检测手段等全新的检测技术。
在全面提升HPLC的速度、灵敏度及分离度的同时,保留其原有的实用性及原理。
基于小颗粒技术的UPLC,并非普通HPLC系统改进而成。
它不但需要耐压、稳定的小颗粒填料(可达1.7µm),而且需要耐压的色谱系统(>15,000psi)、最低交叉污染的快速进样器、快速检测器及优化的系统体积等诸多方面的保障,以充分发挥小颗粒技术优势。
这就需要对系统所有硬件和软件的进行全面创新。
世界第一个商品化UPLC产品是Waters ACQUITY UPLC TM超高效液相色谱系统,它于2004年3月投入市场。
图1:填料技术的沿革1.小颗粒填料改善分离的理论与科学基础液相色谱30年的发展史是颗粒技术的发展史。
颗粒大小的改变直接影响到柱效,从而对分离结果产生直接影响。
由上图可知:随着颗粒度的不断降低,色谱分离度不断提高。
事实上,上述规律的理论基础是著名的Van Deemeter方程。
Van Deemeter方程是色谱科学家预测颗粒度变化而引起的色谱变化的根本依据。
Van Deemeter曲线(见图2)预测最佳柱效与相应的流动相流速。
由Van Deemeter方程得知:随着颗粒度减小,相应的理论塔板高度(HETP)也下降,得到的柱效会更高。
还应该注意到1.7 µm颗粒的HETP最小值区域扩大了(见图2),这表明可以在比大颗粒更宽的流量范围内得到最高的柱效,结果可以不损失高分离度的同时来适当提高流动相的流速(分析速度)。
小颗粒填料为色谱分离带来如此的高柱效和速度优势,使得利用小颗粒技术成为色谱科学家梦寐以求的目标。
然而HPLC系统的设计,一直难于发挥出最小颗粒的优点。
小颗粒技术的运用,不但要求仪器在超出目前限度(6000 psi/ 400 bar)的压力下工作,同时要求仪器系统的死体积要更小,以便不影响梯度性能,而且还要检测器能高速检测出峰宽只有几秒的色谱峰。


超高效液相色谱法在中药分析领域中的应用现状及展望一、本文概述随着科技的不断进步和人们对中药认识的深入,中药分析领域正面临着前所未有的发展机遇。
超高效液相色谱法(UPLC)作为一种先进的色谱分析技术,以其高分辨率、高灵敏度、高分离效能和快速分析等特点,在中药分析领域中的应用日益广泛。
本文旨在综述超高效液相色谱法在中药分析领域的应用现状,探讨其发展前景,为中药的现代化和国际化提供技术支持。
本文将首先介绍超高效液相色谱法的基本原理和优势,阐述其在中药成分分析、质量控制、药物代谢等方面的应用案例。
然后,我们将重点分析超高效液相色谱法在中药分析领域中的优势和挑战,包括其对于复杂中药体系的处理能力、对于痕量成分的检测能力以及在实际应用中可能遇到的问题。
我们将展望超高效液相色谱法在中药分析领域的未来发展,包括技术创新、方法优化、多技术联用等方面,以期推动中药分析技术的不断进步和发展。
二、超高效液相色谱法在中药分析领域的应用现状超高效液相色谱法(UPLC)作为一种先进的色谱分析技术,近年来在中药分析领域得到了广泛应用。
其高效的分离能力和高灵敏度,使得UPLC成为中药复杂成分分析的有力工具。
在中药指纹图谱的构建中,UPLC发挥了关键作用。
通过优化色谱条件和选择适当的检测器,UPLC能够实现对中药中多种成分的快速、准确分离和检测。
这不仅有助于中药质量控制,还可以为中药的药效物质基础研究和质量控制提供科学依据。
UPLC在中药有效成分的分析中也表现出色。
通过精确测量中药中有效成分的保留时间和峰面积,可以实现对中药中有效成分的定量分析。
这为中药的质量评价、药效研究以及新药开发提供了有力支持。
同时,UPLC在中药代谢产物的分析中也有着重要应用。
通过分析中药在体内的代谢产物,可以深入了解中药的药效机制和药代动力学过程。
这对于中药的临床应用和新药研发具有重要意义。
然而,尽管UPLC在中药分析领域的应用取得了显著进展,但仍面临一些挑战。

超高效液相色谱(UPLC)在药物分析领域中的应用摘要:化学化工产业的发展要求化学分析技术和方法必须进行不断更新,高效液相色谱分析方法就是在这种形势下发展起来的一种现代化分析方式,它具有分析速度快、分离效率高等优势,是目前药物分析领域重要的检验手段和分析技术。
超高效液相色谱法(UPLC)是基于高效液相色谱法的基础上形成的药物分析新技术,与高效液相色谱分析方式相比,它增加了高压输液泵、高效固定相、信息化、机械化以及高灵敏度等检测机械,使得化学药物成分分离速度更快、分析效率更高,并且实现了反复测试,因此该方法已被广泛应用于各种药物质量检测中。
关键词:超高液相色谱;药物分析;应用研究随着社会对药物分析的要求越来越高,超高效液相色谱仪逐渐被广泛应用。
超高效液相色谱技术对药物开展分析的领域中具有速度快、精度高、适用范围更广等特点,其测试全自动化,药品成分容易吸收且处理方法简单的特点,同时它拥有高效液相色谱技术的所有优势,这些使超高效液相色谱技术在不同的药物分析领域得到了较高的应用,同时也被不同的行业所广泛认可。
一、制剂药物分析领域的应用在对药物分析的实际工作中,通常可供分析和提取的药物量是非常少的,因此给提取、分离、分析工作带来了很大的难度。
所以,我们迫切需要一个能够提高分析速度,提升分离效率的技术,其应该具有灵敏度高、精确度高的特点。
超高效液相色谱技术在解决这些问题时,就提供了强有力的支持,逐步显示出其具有良好的发展前景。
对Az1、Azz 和A3z这三种化合物的研究分析中,在保证分析效果相同的前提下,使用超高效液相色谱技术可以大大地缩短分析时间,其精度和灵敏度也相比于其他技术要好很多。
超高效液相色谱技术适合用于常规药物的分析,使用混合模式的固相萃取和超高效液相色谱技术同时使用测定相应的药物成分,其最终的分析结果可以达到美国食品和药物管理局在精度和准确度方面的要求,因此该技术在制剂药物分析领域具有着非常光明的应用前景。

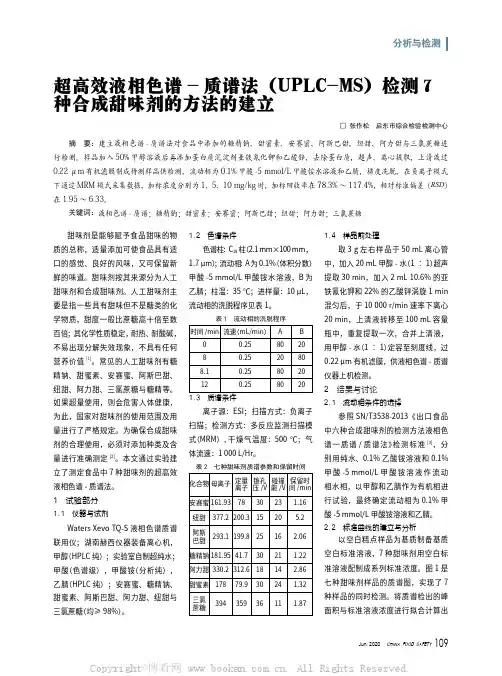
分析与检测甜味剂是能够赋予食品甜味的物质的总称,适量添加可使食品具有适口的感觉、良好的风味,又可保留新鲜的味道。
甜味剂按其来源分为人工甜味剂和合成甜味剂。
人工甜味剂主要是指一些具有甜味但不是糖类的化学物质,甜度一般比蔗糖高十倍至数百倍;其化学性质稳定,耐热、耐酸碱,不易出现分解失效现象,不具有任何营养价值[1]。
常见的人工甜味剂有糖精钠、甜蜜素、安赛蜜、阿斯巴甜、纽甜、阿力甜、三氯蔗糖与糖精等。
如果超量使用,则会危害人体健康,为此,国家对甜味剂的使用范围及用量进行了严格规定。
为确保合成甜味剂的合理使用,必须对添加种类及含量进行准确测定[2]。
本文通过实验建立了测定食品中7种甜味剂的超高效液相色谱-质谱法。
1 试验部分1.1 仪器与试剂Waters Xevo TQ-S液相色谱质谱联用仪;湖南赫西仪器装备离心机,甲醇(HPLC纯);实验室自制超纯水;甲酸(色谱级),甲酸铵(分析纯),乙腈(HPLC纯);安赛蜜、糖精钠、甜蜜素、阿斯巴甜、阿力甜、纽甜与三氯蔗糖(均≥98%)。
1.2 色谱条件色谱柱:C18柱(2.1 mm×100 mm,1.7 μm);流动相:A为0.1%(体积分数)甲酸-5 mmol/L甲酸铵水溶液,B为乙腈;柱温:35 ℃;进样量:10 μL,流动相的洗脱程序见表1。
表1 流动相的洗脱程序时间/min流速(mL/min)A B00.25802080.2520808.10.258020120.2580201.3 质谱条件离子源:ESI;扫描方式:负离子扫描;检测方式:多反应监测扫描模式(MRM),干燥气温度:500 ℃;气体流速:1 000 L/Hr。
表2 七种甜味剂质谱参数和保留时间化合物母离子定量离子锥孔压/V碰撞能/V保留时间/min安赛蜜161.93783023 1.16纽甜377.2200.31520 5.2阿斯 巴甜293.1199.82516 2.06糖精钠181.9541.73021 1.22阿力甜330.2312.61814 2.86甜蜜素17879.93024 1.32三氯 蔗糖3943593611 1.871.4 样品前处理取3 g左右样品于50 mL离心管中,加入20 mL甲醇-水(1∶1)超声提取30 min,加入2 mL 10.6%的亚铁氰化钾和22%的乙酸锌涡旋1 min混匀后,于10 000 r/min速率下离心20 min,上清液转移至100 mL容量瓶中,重复提取一次,合并上清液,用甲醇-水(1∶1)定容至刻度线,过0.22 μm有机滤膜,供液相色谱-质谱仪器上机检测。

过氧化苯甲酰(UPLC)超高效液相色谱法作业指导书过氧化苯甲酰(UPLC)超高效液相色谱法作业指导书1范围本指导书规定了以高效液相色谱测定过氧化苯甲酰的原理、试剂和材料、仪器设备、分析步骤、计算方法和精密度。
本方法最低检出限为0000.5g/kg。
2原理由甲醇提取的过氧化苯甲酰,用碘化钾作为还原剂将其还原为苯甲酸,高效液相色谱分离,在230nm下检测。
3试剂和材料碘化钾:50%水溶液(质量浓度)。
苯甲酸:国家标准物质,纯度≥99.9%。
甲醇:色谱纯。
乙酸铵:称取0.770克乙酸铵用水溶解并稀释至1000ml,混匀后用0.22um的微孔滤膜过滤后使用。
标准溶液:过氧化苯甲酰的标准溶液浓度分别为0ug/ml、5.0ug/ml、10.0ug/ml、20.0ug/ml、40.0ug/ml、80.0ug/ml、100.0ug/ml的标准使用液。
4仪器设备及色谱条件4.1 仪器设备.4.1.1(UPLC)超高效液相色谱仪:配有紫外检测器。
4.1.2 天平:感量0.0001g4.1.3 漩涡混合器4.2 色谱条件4.21 色谱柱:C 18柱、50mmX2.1mm ,1.7um ;和C 18柱、100mmX2.1mm ,1.7um ;4.22 检测波长:230nm 。
4.23 流动相: 乙腈:乙酸铵溶液为10:90(体积分数)。
4.24 流速: 0.3-0.4ml/min 。
4.25 进样量: 3ul 。
5 分析步骤5.1 称取样品2g (精确至0.1mg )与50ml ,具塞比色管中,加10ml 甲醇,在漩涡混合器上混匀1min ,静置5min ,加50%碘化钾水溶液5.0ml ,在漩涡混合器上混匀1min ,放置10min 。
加水至50ml ,混匀,静置。
吸取上层清液通过0.22um 滤膜,待上机分析。
6 计算方法按下式计算过氧化苯甲酰含量:1000(/)0.99210001000c V X g kg X m ⨯⨯=⨯⨯ 式中X----样品中过氧化苯甲酰的含量,单位为克每千克(g/kg );c-----待测样液中过氧化苯甲酰的浓度,单位为微克每毫升(ug/ml ); V-----试样提取液体积,单位为毫升(ml );m-----样品质量,单位为克(g );0.992-----由苯甲酸换算成过氧化苯甲酰的换算系数:242.2/(2x122.1)结果保留两位有效数字。

UPLC(超高效液相色谱)简介超越HPLC随着科学技术的进步,对液相色谱技术的要求也不断提高,单从技术角度的改进已经不行。
这就需要同时从科学与技术的角度出发,或者说从理论高度对液相色谱重新认识。
因此,UPLC(超高效液相色谱)概念得以提出,将HPLC的极限作为自己的起点。
在1996年,Waters公司推出Alliance HPLC时的主要目标是提高液相色谱的"精度"。
当时多数公司都认为HPLC技术已经发展到极致了、而同时用户对性能没有更高的需求,因此HPLC的目标应该是降低成本、走向更低的价格以获得更广泛的应用。
针对这样的观念,Waters公司提出:HPLC的技术没有到达极限,用户对HPLC有更高的要求,HPLC精度的提高对更好、更可靠的结果有极大的益处,对法规的遵从也是一个极大的促进。
站在当今世界科技前沿的液相色谱用户现在又有了新的需求。
首先是改进生产力的需求,因为大量的样品需要在很短的时间内完成,例如代谢组学分析;其次是在生化样品及天然产物样品的分析中,样品的复杂性对分离能力提出了更高的要求;第三是在与MS及MS/MS等检测技术联用时,对连接的质量提出了更高的要求。
简而言之,我们需要"更快地得到更好的结果"。
今天我们发现,随着科学技术的进步,对液相色谱技术的要求也不断提高,单从技术角度的改进已经不行。
这就需要同时从科学与技术的角度出发,或者说从理论高度对液相色谱重新认识。
因此UPLC(超高效液相色谱)概念的提出也就十分自然。
简而言之,UPLC是用HPLC的极限作为自己的起点。
理论基础早在1956年,J.J van Deemter就发表了他著名的理论:van Deemter曲线及其方程式。
最早这个理论是用在气相色谱上的,但是后来出现的液相色谱上也能应用这个理论。
Waters公司引入UPLC的概念就是由研究这个著名的方程式开始。
首先探讨一下这个著名的方程式。

2020年12月邓博等.超高效液相色谱-串联质谱在中西医药品检测与分析中的应用49超高效液相色谱-串联质谱在中西医药品检测与分析中的应用邓博,邓护军,杨飞,门靖西安万隆制药股份有限公司,西安710119摘要超高效液相色谱-串联质谱(UPLC-M S/MS)是一种高效、迅速、稳定性和精密度高的综合性分析技术,其应用前景十分广阔。
本文着重介绍了近年来UPLC- MS/MS在人体化学药品、动物体内化学药品、以及中医药分析与检测三方面的最新应用研究,并展望了其发展前景,以期为医药产品检测与分析、临床药物应用提供参考。
关键词液相色谱串联质谱中西医检测分析应用超高效液相色谱-串联质谱(UPL C - M S/ M S)检测分析技术具有高效分离度和超高灵敏度 的优势,可显著提高数据分析与检测的可靠性与 耐用性,增强目标化合物的分析效率与准确度,是 分析检测领域内的一种综合性的分析手段[3_4]。
近年来,UPLC - M S/M S分析技术在医药[5-6]、化 工[7]、生物工程[84、保健食品[1°]、特种材料"1]等众多领域应用广泛,尤其是化工与医药产品的 检测与残留分析凭借U P L C- M S/M S获得了大幅 度的提升与改进。
本文将重点综述U P L C- M S/M S在中西医药 品检测与分析中的应用研究现状,从人体化学药 品、动物体内化学药品、以及中医药分析与检测三 方面归纳U P L C -M S/M S最新应用研究进展。
1 UPLC-M S/M S应用于人体化学药品的检测1.1 UPLC- M S/M S检测抗菌药物泊沙康唑是一种三唑类广谱抗真菌药物,具 有高效、广谱、低毒等特点[U]。
检测该药物在人 体血浆中的浓度对监测药物吸收、指导临床用药 安全尤为重要[1243]。
金鸿宾等[14]建立了一种特 异性强、灵敏度高的U P L C - M S/M S法测定血浆 中泊沙康唑浓度方法。

基因毒性杂质研究专栏㊀基金项目:中国食品药品检定研究院关键技术研究基金(No.GJJS-2022-4-2)ꎻ#同为第一作者ꎬ∗同为通信作者作者简介:袁松ꎬ男ꎬ硕士ꎬ助理研究员ꎬ研究方向:化学药品质量控制ꎬE-mail:yuansong@nifdc.org.cnꎻ李婕ꎬ女ꎬ硕士ꎬ副主任药师ꎬ研究方向:化学药品质量控制ꎬE-mail:lijie@nifdc.org.cn通信作者:刘阳ꎬ男ꎬ博士ꎬ研究员ꎬ研究方向:药品质量安全ꎬTel:010-53851571ꎬE-mail:yangliu@nifdc.org.cnꎻ张庆生ꎬ男ꎬ硕士ꎬ主任药师ꎬ研究方向:药品质量安全ꎬTel:010-53851375ꎬE-mail:zqs@nifdc.org.cnUPLC-MS/MS法测定磷酸西格列汀中基因毒性杂质NTTP袁松#ꎬ李婕#ꎬ张娜ꎬ张龙浩ꎬ刘阳∗ꎬ张庆生∗(中国食品药品检定研究院ꎬ国家药品监督管理局化学药品质量研究与评价重点实验室ꎬ北京102629)摘要:目的㊀建立磷酸西格列汀原料药及制剂中基因毒性杂质Nitroso-STG-19(NTTP)的超高效液相色谱-串联质谱(UPLC-MS/MS)检测方法ꎮ方法㊀色谱柱为EclipsePlusC18RRHD(3.0mmˑ150mmꎬ1.8μm)ꎬ以含0.1%甲酸的水溶液为流动相Aꎬ0.1%甲酸的乙腈溶液为流动相Bꎬ梯度洗脱ꎬ流速为0.35mL min-1ꎬ柱温为30ħꎬ进样器温度为10ħꎬ进样体积为5μLꎮ采用多反应监测(MRM)模式ꎬ对磷酸西格列汀原料药及制剂中的NTTP进行定量检测ꎮ结果㊀NTTP在0.54~53.93ng mL-1范围内具有良好的线性关系ꎮ检测限和定量限分别为0.02ng mL-1和0.08ng mL-1ꎮ原料药及制剂在低㊁中㊁高3个浓度的平均加样回收率范围(n=3)分别为105.43%~107.21%(RSDɤ3.2%)及115.03%~120.20%(RSDɤ3.6%)ꎮ应用该方法对12批原料药及3批制剂中的NTTP进行检测ꎬ结果显示均有检出ꎮ结论㊀该方法灵敏度高㊁专属性强ꎬ可用于测定磷酸西格列汀原料药及制剂中的NTTPꎬ可为磷酸西格列汀原料药及制剂的质量控制提供参考ꎮ关键词:磷酸西格列汀ꎻ基因毒性杂质ꎻNitroso-STG-19ꎻ含量测定ꎻ超高效液相色谱-串联质谱中图分类号:R927.1㊀文献标志码:A㊀文章编号:2095-5375(2023)12-1000-005doi:10.13506/j.cnki.jpr.2023.12.009DeterminationofgenotoxicimpurityNTTPinSitagliptinPhosphatebyUPLC-MS/MSYUANSong#ꎬLIJie#ꎬZHANGNaꎬZHANGLonghaoꎬLIUYang∗ꎬZHANGQingsheng∗(NMPAKeyLaboratoryforQualityResearchandEvaluationofChemicalDrugsꎬNationalInstitutesforFoodandDrugControlꎬBeijing102629ꎬChina)Abstract:Objective㊀ToestablishanUPLC-MS/MSmethodfordeterminationofgenotoxicimpurityNitroso-STG-19(NTTP)inSitagliptinPhosphate.Methods㊀TheseparationofNTTPwasperformedonaEclipsePlusC18RRHD(3.0mmˑ150mmꎬ1.8μm)withthemobilephaseconsistingof0.1%formicacidaqueoussolution(mobilephaseA)and0.1%formicacidmethanolsolution(mobilephaseB)underagradientelutionataflowrateof0.35mL min-1andacolumntem ̄peratureof30ħ.Multiplereactionmonitoring(MRM)wasperformedonatriplequadrupolemassspectrometerinpositivemode.Results㊀Thecalibrationcurveswasingoodlinearityintherangeof0.54~53.93ng mL-1.Thelimitofdetectionwas0.02ng mL-1ꎬandthelimitofquantificationwas0.08ng mL-1.Therecoveries(n=3)ofactivepharmaceuticalingredients(API)andtabletsatlowꎬmiddleꎬandhighspikedconcentrationswere105.43%~107.21%(RSDɤ3.2%)and115.03%~120.20%(RSDɤ3.6%)respectively.UsingthedevelopedmethodꎬwedetectedNTTPin12batchesofAPIand3batchesofpreparation.TheresultsshowedthatNTTPwasdetectedinallbatches.Conclusion㊀ThemethodwassensitiveandaccurateꎬwhichcanbeappliedforthequantificationsofNTTPinSitagliptinPhosphateꎬprovidingreferenceforqualitycontrolofSita ̄gliptinPhosphate.Keywords:SitagliptinPhosphateꎻGenotoxicimpurityꎻNTTPꎻContentdeterminationꎻUPLC-MS/MS㊀㊀N-亚硝胺类化合物(N-nitrosaminesꎬNAs)是一类结构通式为R1(R2)N-N=O的化合物ꎬ其中R1和R2为烷基或芳烃ꎬ国际癌症研究机构于1987年将NAs列入致癌物清单ꎮ2017年世界卫生组织发布的致癌清单中ꎬ有近16个短脂肪链的N-亚硝胺类化合物被列为2类致癌物质[1]ꎮNAs常常存在于食品㊁饮用水㊁烟草㊁化妆品等物质中[2-5]ꎬ人们长期接触含NAs的这些物质会产生潜在危害ꎮ自2018年欧洲药品管理局(EuropeanMedicinesAgencyꎬEMA)宣布在缬沙坦原料和制剂中检测出N-亚硝基二甲胺(NDMA)后ꎬ各国药品监管机构纷纷加强对药品中NAs的监测ꎬ并对多批含有NDMA或其他亚硝胺类杂质的沙坦类药物进行召回ꎬ并要求对产品中NAs存在进行风险评估及建立合适的控制策略[6-9]ꎮ西格列汀(结构式见图1)是一种二肽基肽酶-4(DPP-4)抑制剂ꎬ可以刺激胰高血糖素的释放并减少胰岛素的分泌ꎬ从而发挥降糖作用ꎬ是一种常见的降糖药ꎬ可与其他药联合用药来治疗包括超重肥胖㊁胰岛素治疗不佳㊁肥胖型2型糖尿病等疾病ꎬ治疗效果良好[10-12]ꎮ2022年报道表明西格列汀合成中间体三氟甲基-5ꎬ6ꎬ7ꎬ8-四氢-1ꎬ2ꎬ4-三唑并-[4ꎬ3-α]-吡嗪盐酸盐(TTP)[13]可能与亚硝酸盐反应生成Nitroso-STG-19(NTTPꎬ见图1)ꎮNTTP是新发现的一种存在于西格列汀中的基因毒性杂质ꎬ可能是以TTP为原料合成西格列汀时产生或是最终产品中残留的TTP与辅料中微量的亚硝酸盐反应生成NTTPꎮEMA和美国食品药品监督管理局(FDA)最初都将其最大摄入量设定为37ng d-1ꎬ但FDA为了避免西格列汀产品的短缺ꎬ将最大摄入量调整为246.7ng d-1ꎬ暂未召回该产品[14-15]ꎮ目前国内外尚无对西格列汀中的NTTP检测方法的报道ꎬ本文建立了一个超高效液相色谱-串联质谱(UPLC-MS/MS)检测西格列汀中NTTP的方法ꎬ为西格列汀相关原料药与制剂的药品质量控制和监管提供参考ꎮ图1㊀西格列汀㊁TTP和NTTP的结构式1㊀材料1.1㊀仪器㊀高效液相色谱-串联三重四极杆质谱仪(美国Waters公司)ꎬ配备电喷雾离子源(ESI)和MassLynxV4.2数据处理系统ꎻXP205DR型电子分析天平(瑞士Mettler公司)ꎻMilli-Q超纯水纯化系统(美国Millipore公司)ꎮ1.2㊀药物与试剂㊀乙腈㊁甲酸㊁甲酸铵(质谱级ꎬ美国FisherScientific)ꎬ水为超纯水ꎬ对照品NTTP来自本实验室合成ꎬ采用高分辨质谱(见图2A)和核磁(见图2B㊁C)对其结构进行了确证ꎮ2㊀方法与结果2.1㊀溶液的制备2.1.1㊀标准曲线溶液的制备㊀取NTTP对照品约10mgꎬ精密称定ꎬ置100mL量瓶中ꎬ加水使溶解并稀释至刻度ꎬ作为对照品储备液ꎻ精密量取对照品储备液适量ꎬ以水为稀释剂逐级定量稀释制成每1mL中含NTTP0.54㊁1.08㊁4.31㊁10.79㊁26.97㊁53.93ng的溶液ꎬ作为系列线性溶液ꎮ2.1.2㊀供试品溶液的制备㊀取磷酸西格列汀原料药约100mgꎬ精密称定ꎬ置10mL量瓶中ꎬ加水适量ꎬ超声使溶解ꎬ用水稀释至刻度ꎬ摇匀ꎬ作为原料药的供试品溶液ꎮ取磷酸西格列汀片10片ꎬ精密称定ꎬ研细ꎬ混匀ꎬ精密称取约相当于磷酸西格列汀100mg的细粉ꎬ置15mL离心管中ꎬ精密加入水10mLꎬ超声并振摇10minꎬ滤过ꎬ取续滤液作为片剂的供试品溶液ꎮ2.2㊀色谱及质谱条件2.2.1㊀色谱条件㊀采用AgilentEclipsePlusC18RRHD(3.0mmˑ150mmꎬ1.8μm)色谱柱ꎬ以含0.1%甲酸的水溶液为流动相Aꎬ0.1%甲酸的乙腈溶液为流动相Bꎬ梯度洗脱:0~8.0minꎬ40%Bң100%Bꎻ8.0~8.1minꎬ100%Bң40%Bꎻ8.1~12.0minꎬ40%Bꎻ流速为0.35mL min-1ꎬ柱温为30ħꎬ进样器温度为10ħꎬ进样体积为5μLꎮ2.2.2㊀质谱条件㊀采用电喷雾离子源(ElectrosprayIonizationSourceꎬESI)ꎬ正离子检测模式ꎬ毛细管电压为4.0kVꎬ脱溶剂气温度为500ħꎬ脱溶剂气流量为1000L h-1ꎬ锥孔气流量为150L h-1ꎬ离子源温度为150ħꎮ监测模式为多反应监测(Multiplere ̄actionmonitorꎬMRM)ꎬ以m/z221.83ң191.95作为定量离子对ꎬ锥孔电压为10Vꎬ碰撞能量为10Vꎬ以离子对m/z221.83ң164.71作为定性离子对ꎬ锥孔电压为10Vꎬ碰撞能量为20Vꎮ采集时间为2.3~8.0minꎮ2.3㊀方法学考察2.3.1㊀专属性试验㊀取水作为空白溶剂和 2.1.1项下约4ng mL-1的对照品溶液分别进样ꎬ记录色谱图ꎬ在所建立的色谱和质谱条件下ꎬNTTP的保留时间为2.62min(见图3A)ꎬ峰型良好ꎬ空白溶剂对检测无干扰ꎮ图2㊀结构确证-高分辨质谱图(AꎬH-ESI+)㊁核磁共振氢谱图(B)和碳谱图(C)2.3.2㊀系统精密度㊀取 2.1.1 项下约4ng mL-1的线性溶液连续进样6次ꎬ得NTTP峰面积的RSD为1.13%ꎬ结果表明系统精密度良好ꎮ2.3.3㊀重复性㊀2.3.3.1㊀原料药重复性㊀取磷酸西格列汀原料药(批号:SGZ1040006)ꎬ按 2.1.2 项下方法平行制备6份原料药供试品溶液ꎬ进样检测ꎬ按标准曲线法计算NTTP含量ꎬ计算得6份原料药供试品溶液中NTTP含量的RSD为7.2%ꎮ结果表明方法对原料药测定具有良好的重复性ꎮ2.3.3.2㊀片剂重复性㊀取磷酸西格列汀片ꎬ按 2.1.2 项下方法平行制备6份片剂供试品溶液ꎬ进样检测ꎬ按标准曲线法计算NTTP含量ꎬ计算得6份片剂供试品溶液中NTTP含量的RSD为3.3%ꎬ结果表明方法对片剂测定具有良好的重复性ꎮ2.3.4㊀线性㊀分别取 2.1.1 项下系列线性溶液ꎬ进样检测ꎬ记录色谱图ꎮ以质量浓度为横坐标(X)ꎬNTTP峰面积为纵坐标(Y)进行线性回归ꎬ在0.54~53.93ng mL-1浓度范围内线性结果为Y=5100.84X+539.79(r=1.0000)ꎮ结果显示NTTP在其线性范围内峰面积与进样浓度之间呈良好的线性关系ꎮ2.3.5㊀回收率试验2.3.5.1㊀原料药回收率试验㊀取磷酸西格列汀原料药(批号:GZ1040006)约100mgꎬ精密称定ꎬ置10mL量瓶中ꎬ分别用 2.1.1 项下25ng mL-1(高浓度)㊁10ng mL-1(中浓度)㊁4ng mL-1(低浓度)的线性溶液溶解并稀释至刻度ꎬ摇匀ꎬ作为原料药回收率溶液ꎬ每个浓度点平行制备3份ꎬ进样检测ꎬ结果见表1ꎬ低㊁中㊁高浓度点的回收率分别为107.21%(RSD2.9%ꎬn=3)㊁105.95%(RSD2.5%ꎬn=3)㊁105.43%(RSD3.2%ꎬn=3)ꎬ结果表明原料药回收率良好ꎮ2.3.5.2㊀片剂回收率试验㊀取磷酸西格列汀片(批号:200515JA)ꎬ精密称定ꎬ研细ꎬ混匀ꎬ精密称取细粉适量(约相当于西格列汀100mg)ꎬ置10mL量瓶中ꎬ分别用 2.1.1 项下25ng mL-1(高浓度)㊁10ng mL-1(中浓度)㊁4ng mL-1(低浓度)的线性溶液溶解并稀释至刻度ꎬ摇匀ꎬ滤过ꎬ取续滤液作为片剂回收率溶液ꎬ每个浓度点平行制备3份ꎬ进样检测ꎬA.空白溶剂ꎻB.对照品溶液ꎻC.片剂供试品溶液图3㊀空白溶剂㊁对照品溶液和片剂供试品溶液提取的离子流色谱图结果见表3ꎬ低㊁中㊁高浓度点的回收率分别为115.03%(RSD2.1%ꎬn=3)㊁115.95%(RSD3.6%ꎬn=3)㊁120.20%(RSD1.4%ꎬn=3)ꎬ结果表明片剂回收率在可接受范围内ꎮ表1㊀原料药加样回收率结果编号加入量/ng检测量/ng本底/ng回收率(%)平均回收率(%)RSD(%)143.1548.722.80106.42243.1547.972.84104.59107.212.9343.1550.632.90110.614107.87117.312.96106.015107.87119.992.85108.59105.952.56107.87114.182.82103.247269.66297.642.84109.328269.66282.852.83103.84105.433.29269.66281.093.01103.122.3.6㊀检测限与定量限㊀取 2.1.1 项下约0.5ng mL-1的线性溶液ꎬ以水为稀释剂逐步稀释ꎬ分别在信噪比为3ʒ1和10ʒ1时作为检测限和定量限ꎬ测得NTTP的检测限和定量限分别为0.02ng mL-1和0.08ng mL-1ꎮ2.3.7㊀提取效率考察㊀取磷酸西格列汀原料药(批号:SGZ1040006)和磷酸西格列汀片(批号:T024447)ꎬ分别按 2.1.2 项平行制备2份溶液ꎬ分别超声并振摇10㊁20minꎬ进样检测ꎬ按标准曲线法计算NTTP含量ꎬ计算得原料药2份供试品溶液中NTTP含量相对标对偏差为3.32%ꎬ片剂2份供试品溶液中NTTP含量相对标对偏差为2.02%ꎬ结果表明超声并振摇10min可提取完全ꎮ表2㊀片剂加样回收率结果编号加入量/ng检测量/ng本底/ng回收率(%)平均回收率(%)RSD(%)143.1550.580117.22243.1549.840115.50115.032.1343.1548.490112.384107.87128.890119.495107.87120.180111.41115.953.66107.87126.150116.957269.66328.640121.878269.66324.350120.28120.201.49269.66319.390118.442.3.8㊀溶液稳定性㊀取 2.1.1 项下约1ng mL-1的对照品溶液ꎬ放置于进样盘ꎬ间隔6h进样检测ꎮ0h和6h时NTTP峰面积的相对偏差为2.77%ꎬ结果表明对照品溶液10ħ时6h内稳定ꎮ2.4㊀样品测定㊀按 2.1 项下制备磷酸西格列汀供试品溶液ꎬ照 2.2 项下条件进样检测ꎬ记录色谱图(典型图谱见图2B㊁C)ꎬ以峰面积按标准曲线法计算供试品溶液中NTTP的含量ꎮ4家原料厂家的12批样品中检出NTTP含量为0.014~0.20μg g-1ꎻ有3批磷酸西格列汀片中检测出NTTPꎬ含量为1.26~1.41μg g-1ꎮ3㊀讨论NTTP在水中易溶ꎬ以水为溶剂配制浓度约为1μg mL-1的对照品溶液ꎬ分别采用ESI离子源和APCI离子源下针泵进样对NTTP的响应及定量/定性离子进行考察ꎬ结果表明NTTP在ESI离子源正离子采集模式下响应更好ꎬ继续对ESI+条件进行优化ꎬ最终确定NTTP定量离子对为m/z221.83ң191.95ꎬ锥孔电压为10Vꎬ碰撞能量为10eVꎬ定性离子对为m/z221.83ң164.71作为ꎬ锥孔电压为10Vꎬ碰撞能量为20eVꎮ以水为溶剂配制含磷酸西格列汀和NTTP浓度均约为100μg mL-1的混合溶液ꎬ采用紫外检测器对NTTP峰与西格列汀峰之间的分离情况进行了考察ꎬ分别试验了甲醇-水㊁乙腈-水系统ꎬ试验证明当流动相为乙腈-水时NTTP和西格列汀出峰较快ꎬ且可完全分离ꎬ水中加入甲酸铵后NTTP的质谱响应降低明显且峰型变差ꎬ加入甲酸可以增强离子化效率提高质谱响应ꎮ最终以含0.1%甲酸的水溶液为流动相Aꎬ0.1%甲酸的乙腈溶液为流动相Bꎬ并进行梯度洗脱ꎮ在最终确定的色谱条件下ꎬ磷酸西格列汀在2.2min即可洗脱完全ꎬ质谱采集时间设定为2.3~8.0minꎬ高浓度的磷酸西格列汀经液相洗脱后直接从废液排出ꎬ以避免对质谱的污染ꎮ磷酸西格列汀片未明确最大单日剂量ꎬ按推荐剂量100mg d-1计ꎻEMA和FDA最初都将NTTP最大摄入量设定为37ng d-1ꎬ但FDA为了避免西格列汀产品的短缺ꎬ将NTTP最大摄入量调整为246.7ng d-1[15]ꎻ如按37ng d-1计算ꎬ则磷酸西格列汀中NTTP的残留限度为0.37μg g-1ꎬ所检原料药中NTTP均未超过此限度ꎬ但片剂中NTTP含量已超限度值ꎻ如按246.7ng d-1计算ꎬ则磷酸西格列汀中NTTP的残留限度为2.47μg g-1ꎬ原料药和片剂中NTTP均未超出此限度ꎮ片剂中NTTP含量显著高于原料药ꎬ可能是由于终产品中的TTP残留继续与极微量的亚硝酸盐继续反应生产NTTPꎬ关于TTP残留量与NTTP残留量之间的相关性还需进行进一步的研究ꎮ4㊀结论本研究建立了磷酸西格列汀原料药及片剂中NTTP的UPLC-MS/MS检测方法ꎬ并进行了相关方法学验证ꎬ结果表明ꎬ所建立的方法具有良好的专属性㊁灵敏度和准确度ꎬ可准确测定磷酸西格列汀原料药及片剂中潜在的NTTP含量ꎬ有助于磷酸西格列汀的市场监管ꎬ保障药品质量安全ꎮ参考文献:[1]㊀袁松ꎬ黄海伟ꎬ于颖洁ꎬ等.UPLC-MS/MS法同时测定氯沙坦钾和缬沙坦中7个亚硝胺类基因毒性杂质[J].药物分析杂志ꎬ2021ꎬ41(7):1218-1225.[2]PARKJEꎬSEOJEꎬLEEJYꎬetal.DistributionofsevenN-NitrosaminesinFood[J].ToxicolResꎬ2015ꎬ31(3):279-298. [3]KRASNERSWꎬMITCHWAꎬMCCURRYDLꎬetal.For ̄mationꎬprecursorsꎬcontrolꎬandoccurrenceofnitrosaminesindrinkingwater:areview[J].WaterResꎬ2013ꎬ47(13):4433-4450.[4]EDWARDSSHꎬHASSINKMDꎬTAYLORKMꎬetal.To ̄bacco-SpecificNitrosaminesintheTobaccoandMain ̄streamSmokeofCommercialLittleCigars[J].ChemResToxicolꎬ2021ꎬ34(4):1034-1045.[5]ALHOOSHANIK.DeterminationofnitrosaminesinskincarecosmeticsusingCe-SBA-15basedstirbar-supportedmicro-solid-phaseextractioncoupledwithgaschromatographymassspectrometry[J].ArabJChemꎬ2020ꎬ13(1):2508-2516.[6]MALIHIFꎬWANGT.Animprovedanalyticalmethodforquantitationofnitrosamineimpuritiesinophthalmicsolu ̄tionsusingliquidchromatographywithtandemmassspec ̄trometry[J].JChromatogrOpenꎬ2022(2):100037. [7]FDA.UpdatesonAngiotensinIIReceptorBlocker(ARB)RecallsIncludingValsartanꎬLosartanandIrbesartan[EB/OL].[2022-12-24].http://www.fda.gov/Drugs/Drug ̄Safety/ucm613916.htm.[8]BHARATESS.CriticalAnalysisofDrugProductRecallsduetoNitrosamineImpurities[J].JMedChemꎬ2021ꎬ64(6):2923-2936.[9]GUNASEKARANPMꎬCHERTOWGMꎬBHALLAVꎬetal.CurrentStatusofAngiotensinReceptorBlockerRecalls[J].Hypertensionꎬ2019ꎬ74(6):1275-1278.[10]HERMANGAꎬSTEVENSCꎬVANDYCKKꎬetal.Phar ̄macokineticsandpharmacodynamicsofsitagliptinꎬanin ̄hibitorofdipeptidylpeptidaseIVꎬinhealthysubjects:Re ̄sultsfromtworandomizedꎬdouble-blindꎬplacebo-controlledstudieswithsingleoraldoses[J].ClinPharmacolTherꎬ2005ꎬ78(6):675-688.[11]HERMANGAꎬBERGMANAꎬLIUFꎬetal.Pharmacoki ̄neticsandpharmacodynamiceffectsoftheoralDPP-4in ̄hibitorsitagliptininmiddle-agedobesesubjects[J].JClinPharmacolꎬ2006ꎬ46(8):876-886.[12]JANANILꎬBAMEHRHꎬTANHAKꎬetal.EffectsofSita ̄gliptinasMonotherapyandAdd-OntoMetforminonWeightLossamongOverweightandObesePatientswithType2Diabetes:ASystematicReviewandMeta-Analysis[J].DrugRes(Stuttg)ꎬ2021ꎬ71(9):477-488.[13]HANSENKBꎬHSIAOYꎬXUFꎬetal.Highlyefficientasymmetricsynthesisofsitagliptin[J].JAmChemSocꎬ2009ꎬ131(25):8598-8804.[14]EuropeanMedicinesAgency.Questionsandanswersformarketingauthorizationholders/applicantsontheCHMPOpinionfortheArticle5(3)ofRegulation(EC)No726/2004referralonnitrosamineimpuritiesinhumanmedicinalproducts[EB/OL].[2022-12-24].https://www.ema.europa.eu/documents/referral/nitrosamines-emea-h-a53-1490-questions-answers-marketing-auth ̄orisation-holders/applicants-chmp-opinion-article-53-regulation-ec-no-726/2004-referral-nitrosamine-impu ̄rities-human-medicinal-products_en.pdf.[15]U.S.FoodandDrugAdministration.QFDAworkstoavoidshortageofsitagliptinfollowingdetectionofnitrosamineim ̄purity[EB/OL].[2022-12-24].https://www.fda.gov/drugs/drug-safety-and-availability/fda-works-avoid-shortage-sita ̄gliptin-following-detection-nitrosamine-impurity.(收稿日期:2023-02-25)。
张朋翠 1 李述溪 21.青岛黄海制药有限责任公司山东青岛2660002.浙江普利药业有限公司浙江杭州310000摘要:本文主要目的就是对超高效液相色谱法(UPLC,Ultra Performance Liquid Chromatography)在药物检验中的应用效果进行分析。
采用的方法就是,选择利福平、吡嗪酰胺与异烟肼样品,以高效液相色谱法(HPLC,High Performance Liquid Chromatography)方法为对照组,以UPLC为研究组,对比相应检验的方法。
发现,相比于HPLC方法的分析时间,UPLC方法的分析时间更短,而检验结果的差异具有统计学意义。
最终确定,借助UPLC方法能够充分提高检验效率。
关键词:药物检验;UPLC;研究分析前言:UPLC主要是基于HPLC发展而来的技术。
检测效率非常突出,并增加了色谱峰容量、灵敏度以及分析通量等优势,同时在液相实验中得到广泛应用。
基于社会快速发展以及民众生活水平提升过程中,民众在药物、食品质量等方面要求不断提升,相关分离解析技术得到快速更新与发展。
UPLC能够快速检测样品中各种组分,便捷地处理样品,进行自动化检测,具有广泛应用范围,可以快速分离,为药品监测以及食品质量安全等方面提供良好检测方法[1]。
1方法与资料1.1资料样品选择常州制药公司生产的利福平、成都锦华公司生产的吡嗪酰胺、沈阳红旗公司生产的异烟肼,结合相关检验方法设计研究组与对照组,同时比较两组资料,P>0.05差异没有统计学意义。
1.2方法对照组选择HPLC方法开展检测工作,具体操作如下:选择10g利福平、10g吡嗪酰胺、10g异烟肼,将样品磨成粉末,将适量粉末装入500ml量瓶中,在溶解、稀释以及过滤等操作之后,在200—400mm紫外波长条件进行扫描(在210mm波长进行测定,选择Waters Sunfire C18色谱柱,规格为:5um,250×4.6mm;以乙腈-0.075mol/L磷酸二氢钾溶液(5:5)为流动相,取样品溶液(20ul),注入HPLC中,并记录色谱图[2]。
色谱分析(中国药科大学)超高效液相色谱(UPLC)超高效液相色谱(UPLC)超高效液相色谱技术(ultra performance liquid chcromatography,简称UPLC)是一种综合了小颗粒填料、非常低系统体积(死体积)及快速检测手段等全新的检测技术。
在全面提升HPLC的速度、灵敏度及分离度的同时,保留其原有的实用性及原理。
基于小颗粒技术的UPLC,并非普通HPLC系统改进而成。
它不但需要耐压、稳定的小颗粒填料(可达 1.7µm),而且需要耐压的色谱系统(>15,000psi)、最低交叉污染的快速进样器、快速检测器及优化的系统体积等诸多方面的保障,以充分发挥小颗粒技术优势。
这就需要对系统所有硬件和软件的进行全面创新。
世界第一个商品化UPLC产品是Waters ACQUITY UPLC TM超高效液相色谱系统,它于2004年3月投入市场。
图1:填料技术的沿革1.小颗粒填料改善分离的理论与科学基础液相色谱30年的发展史是颗粒技术的发展史。
颗粒大小的改变直接影响到柱效,从而对分离结果产生直接影响。
由上图可知:随着颗粒度的不断降低,色谱分离度不断提高。
事实上,上述规律的理论基础是著名的Van Deemeter方程。
Van Deemeter方程是色谱科学家预测颗粒度变化而引起的色谱变化的根本依据。
Van Deemeter曲线(见图2)预测最佳柱效与相应的流动相流速。
由Van Deemeter 方程得知:随着颗粒度减小,相应的理论塔板高度(HETP)也下降,得到的柱效会更高。
还应该注意到1.7 µm颗粒的HETP最小值区域扩大了(见图2),这2表明可以在比大颗粒更宽的流量范围内得到最高的柱效,结果可以不损失高分离度的同时来适当提高流动相的流速(分析速度)。
小颗粒填料为色谱分离带来如此的高柱效和速度优势,使得利用小颗粒技术成为色谱科学家梦寐以求的目标。
然而HPLC系统的设计,一直难于发挥出最小颗粒的优点。
超高效液相色谱(UPLC)
超高效液相色谱技术(ultra performance liquid chcromatography,简称UPLC)是一种综合了小颗粒填料、非常低系统体积(死体积)及快速检测手段等全新的检测技术。
在全面提升HPLC的速度、灵敏度及分离度的同时,保留其原有的实用性及原理。
基于小颗粒技术的UPLC,并非普通HPLC系统改进而成。
它不但需要耐压、稳定的小颗粒填料(可达1.7µm),而且需要耐压的色谱系统(>15,000psi)、最低交叉污染的快速进样器、快速检测器及优化的系统体积等诸多方面的保障,以充分发挥小颗粒技术优势。
这就需要对系统所有硬件和软件的进行全面创新。
世界第一个商品化UPLC产品是Waters ACQUITY UPLC TM超高效液相色谱系统,它于2004年3月投入市场。
图1:填料技术的沿革
1.小颗粒填料改善分离的理论与科学基础
液相色谱30年的发展史是颗粒技术的发展史。
颗粒大小的改变直接影响到柱效,从而对分离结果产生直接影响。
由上图可知:随着颗粒度的不断降低,色谱分
离度不断提高。
事实上,上述规律的理论基础是著名的Van Deemeter方程。
Van Deemeter方程是色谱科学家预测颗粒度变化而引起的色谱变化的根本依据。
Van Deemeter曲线(见图2)预测最佳柱效与相应的流动相流速。
由Van Deemeter方程得知:随着颗粒度减小,相应的理论塔板高度(HETP)也下降,得到的柱效会更高。
还应该注意到1.7 µm颗粒的HETP最小值区域扩大了(见图2),这表明可以在比大颗粒更宽的流量范围内得到最高的柱效,结果可以不损失高分离度的同时来适当提高流动相的流速(分析速度)。
小颗粒填料为色谱分离带来如此的高柱效和速度优势,使得利用小颗粒技术成为色谱科学家梦寐以求的目标。
然而HPLC系统的设计,一直难于发挥出最小颗粒的优点。
小颗粒技术的运用,不但要求仪器在超出目前限度(6000 psi/ 400 bar)的压力下工作,同时要求仪器系统的死体积要更小,以便不影响梯度性能,而且还要检测器能高速检测出峰宽只有几秒的色谱峰。
在利用杂化颗粒技术合成出耐压的新一代小颗粒色谱填料之后,UPLC超高效液相色谱系统的设计,充分利用了小颗粒填料的所有优点,弥补传统HPLC系统的不足。
图2: 范德米特(van Deemeter)曲线
1.1 超高分离度
根据等度液相色谱分离的分离度(Rs)方程,分离度(Rs)与柱效(N)的平方根成正
比。
N 4
)(α-1α)k 2(k 2+1)(1)
按Van Deemter 色谱理论,柱效(N)与颗粒度(dp)成反比:
L dp ∝N (2)
故:随着dp 的降低,N 值会增加;而N 值增加,则Rs 值增加。
HPLC 与UPLC
的基本分离理论,进一步说明了颗粒度大小和分离度密不可分的关系。
新一代UPLC系统发挥了1.7 µm颗粒提供柱效增高的全部优越性。
尤其是1.7 µm颗粒提供的柱效比5 µm颗粒提高了3倍。
因为分离度与粒度的平方根成反比,1.7 µm颗粒的分离度比5 µm颗粒提高了70%。
在梯度分离中也具有同样的优越性,此时分离能力用峰容量衡量。
UPLC用1.7 µm颗粒提高了分离能力,可以分离出更多的色谱峰(见图3),从而对样品提供的信息达到了一个新的水平。
而且又最大地缩短了开发方法所需的时间。
图3显示UPLC可以大大提高分离度,同时色谱峰强度也得到了提高。
图3: UPLC与HPLC:分离度比较
1. 2 超高速度
较小的颗粒能超乎寻常地提高分析速度而不降低分离度。
因为颗粒度减小后,柱长可以按比例缩短而保持柱效不变(见式3),而且Van Deemter理论表明最佳流速
与粒度成反比(见式4)。
柱长缩短会加快分离速度,而颗粒度越小,最佳流速也越大,进而可以通过提高流速来进一步加快分离速度。
(3)
由于新一代UPLC 系统用1.7 µm 颗粒,柱长可以比用5 µm 颗粒时缩短3倍而
保持柱效不变,而且使分离在高3倍的流速下进行,结果使分离过程快了9倍而分离度保持不变。
1.3 超高灵敏度
过去几年中,提高灵敏度的工作集中在检测器上,包括光学检测器和质谱检测
器。
这种趋势主要是受要求检测化合物的浓度越来越低(如高效药物)的驱动。
然而采用超高性能色谱系统就能获得灵敏度的显著提高。
在UPLC 中始终可得到较高的灵敏度。
UPLC 使用小颗粒技术可以得到更高的
柱效(因而改善了分离度)、更窄的色谱峰宽(见式5),即更高的灵敏度。
因为色谱峰变得更窄,峰高也就更高了(见式6);同样,当UPLC 用于快速分析、
用较短色谱柱而使柱效不变时,色谱峰高会相应增加 (见式7)。
因此,使用UPLC 技术,不仅可以在保持与HPLC 相同分离度时提高峰高,而且在改善分离度的同时亦可提高峰高即灵敏度。
1
最佳流速∝ ——……..(4) dp 1 峰高∝——…..(6) W 1
峰高∝——……(7) L
图5: HPLC到UPLC:灵敏度的改善无需折衷
1.4 UPLC为最佳的质谱入口
UPLC与质谱联用,可以实质性地改善质谱检测结果的质量。
UPLC的特殊性能使质谱检测器的性能首次得以充分体现。
由于低流速下色谱峰扩散不大,增加了峰浓度,有利于提高离子源的效率,因而使灵敏度至少提高了3倍。
除UPLC技术本身带来的速度、灵敏度和分离度的改善外,UPLC的超强分离能力有助于待测物与同其竞争电离的杂质的分离,从而可以使质谱检测器的灵敏度因离子抑制现象的减弱或克服而得到进一步的提高。
故使用UPLC-MS联用,可以获得灵敏度较HPLC-MS联用系统大有改善的分离结果,获得更多、质量更好的信息。
图6: HPLC-MS到UPLC-MS:灵敏度的额外提高
1.5简单方便的方法转换
UPLC与HPLC基于相同的分离机理,故相互之间的方法转换非常容易和方便。
现有HPLC方法可以按照比例直接转换成UPLC方法;相反,UPLC方法也很容易可以转换成HPLC方法供常规HPLC系统使用。
UPLC不仅比传统HPLC具有更高的分离能力,而且结束了人们多年不得不在速度和分离度之间取舍的历史。
使用UPLC可以在很宽的线速度、流速和反压下进行高效的分离工作,并获得优异的结果。
2 UPLC的创新和技术基础1
高柱效的UPLC色谱柱是UPLC技术中最重要的部分。
新一代应用“杂化颗粒技术”(Hybrid particle technology,HPT)研制的桥式乙烷-硅碳杂化颗粒具有耐高压,宽pH使用范围(pH 2~12)的特点。
Waters公司为UPLC色谱柱装备了一台用独立柱塞驱动,能进行4种溶剂切换的二元高压梯度输液泵。
耐压可达16000psi,在这个压力下,溶剂尤其是梯度分离时使用的混合溶剂,其压缩性会有显著变化,因此溶剂输送系统可在很宽压力范围内,具有补偿溶剂压缩性变化的能力。
从而能在等度或者梯度条件下保持流速的稳定性和梯度的重现性。
在UPLC中为保护色谱柱不受极端高压力波动的影响,进样过程应当相对无压力波动;进样系统的死体积应足够小以降低样品谱带展宽;快速进样周期可以使得UPLC在具有高样品容量的同时也实现高速度,还具有极低交叉污染的小体积进样的能力。
Acquity UPLC TM采用的进样新技术包括:针内针进样探头(XYZZ’),它是一种高速进样机械装置,它是使用液相色谱管路(PEEK材料)充当进样针以减少死体积,而“外针”是一小段硬管,用来扎破样品瓶塞;压力辅助进样,采用一强一弱的双溶剂清洗进样针以降低进样时候的交叉污染。
UPLC对于检测器有两个要求:高的采样速度,使得它可以收集足够多的数据点,以获得准确,可重现的保留时间和峰面积;检测池的死体积要尽可能小,减少谱带扩展以保持高柱效。
Waters公司的Acquity UPLC TM使用采样速度为40点/s,池体积仅为500 nl(约为HPLC的1/20)的新型光导纤维传导的流通池。
当光束通过光导纤维传入流通池后,利用聚四氟乙烯池壁的全反射特征,不损失光能量,而使得检测灵敏度比HPLC高2~3倍。
光源可使用可变波长的紫外光或二极管阵列系统。
3 UPLC的缺陷
UPLC可以更快的速度和更高的质量完成以往HPLC的工作,为用户节省宝贵的时间和日常溶剂消耗,从而获得最大的投资回报。
但是小颗粒填料在带来高柱效
高分离速度的同时也会引起高的反压,使得工作柱压比普通HPLC高许多,导致UPLC的色谱柱的寿命比较短,这就造成其使用成本过高。
4 UPLC的应用举例
居文政等(2)建立了用UPLC-MS法研究灯盏花乙素在胃肠道的代谢物的方法,确定了以总苷元为检测对象研究灯盏花乙素药代动力学是合理的。
参考文献
1 于士林高效液相色谱方法与应用(第二版)化学工业出版社185-194
2 居文政,储继红,谭仁祥等UPLC-MS/MS联用法分析灯盏花乙素在胃肠道的代
谢物[J]. 中国临床药理学与治疗学,2006;11(3):292-295。