质粒测序引物设计
- 格式:doc
- 大小:7.03 KB
- 文档页数:4

引物设计原则及酶切位点选择和设计:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用[整理]载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来T连入质粒中的重要目的酶切,所以感到还是直接扩增好一点。
但这就需要你仔细设计引物。
扩增出靶基因的时候在核就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR可以在质粒的图谱说明书上酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,找取相应的位点,进行设计。
(一)设计引物前应做的准备工作:准备载体图谱,大致准备把片断插在那个部分对片断进行酶切分析,确定一下那些酶切位点不能用准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题往往导致两个,所连接片断上没有这两个位点,且距离不能太近,两个位点应是载体上的,除非恰好是与上面两个酶在一起的酶切位点。
只能切一个,酶都切不好。
因此,紧挨在一起,还有一种情况是:不能有碱基的交叉,比如promega的说明书上说,最好隔四个。
我看AGATCTTAAG,这样的位点比较难切。
两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。
最好使用酶切效率高的。
的酶。
最好使用双酶切有共同buffer最好使用自己实验室有的酶,这样可,ecor1等),最好使用较常用的酶(如hind3,bamh1以省钱。
的问题,很多的战友都有疑惑。
其实园子里有很多的解释了。
的计算,关于TmTm大家可以理解,双链溶解所需的温度。
即是DNA叫溶解温度(melting temperature, Tm),Tm因此,的溶解是没有作用的。
而不互补的区域对DNA 这个温度是由互补的DNA区域决定的,(除时,只计算互补的区域Tm才有贡献。
计算Tm只有和模板互补的区域对对于引物的Tm,过低,是因为他们误把保护碱。
不少战友设计的引物都Tm 非你的酶切位点也与模板互补)反应的诸多困难。

引物设计原则及酶切位点选择和设计[整理]:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用T载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来酶切,所以感到还是直接扩增好一点。
但这就需要你仔细设计引物。
连入质粒中的重要目的就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR扩增出靶基因的时候在核酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,可以在质粒的图谱说明书上找取相应的位点,进行设计。
(一)设计引物前应做的准备工作:准备载体图谱,大致准备把片断插在那个部分对片断进行酶切分析,确定一下那些酶切位点不能用准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。
因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。
我看promega的说明书上说,最好隔四个。
还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。
两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。
最好使用酶切效率高的。
最好使用双酶切有共同buffer的酶。
最好使用较常用的酶(如hind3,bamh1,ecor1等),最好使用自己实验室有的酶,这样可以省钱。
Tm的计算,关于Tm的问题,很多的战友都有疑惑。
其实园子里有很多的解释了。
Tm叫溶解温度(melting temperature, Tm),即是DNA双链溶解所需的温度。
大家可以理解,这个温度是由互补的DNA区域决定的,而不互补的区域对DNA的溶解是没有作用的。
因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。
计算Tm时,只计算互补的区域(除非你的酶切位点也与模板互补)。
不少战友设计的引物都Tm过低,是因为他们误把保护碱基和酶切位点都计算到Tm里了,最后的结果是导致了PCR反应的诸多困难。

引物设计原则及酶切位点选择和设计[整理]:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用T载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来酶切,所以感到还是直接扩增好一点。
但这就需要你仔细设计引物。
连入质粒中的重要目的就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR扩增出靶基因的时候在核酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,可以在质粒的图谱说明书上找取相应的位点,进行设计。
(一)设计引物前应做的准备工作:准备载体图谱,大致准备把片断插在那个部分准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。
因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。
我看promega的说明书上说,最好隔四个。
还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。
两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。
最好使用酶切效率高的。
最好使用双酶切有共同buffer的酶。
最好使用较常用的酶(如hind3, bamhl, ecorl等),最好使用自己实验室有的酶,这样可以省钱。
Tm的计算,关于Tm的问题,很多的战友都有疑惑。
其实园子里有很多的解释了。
Tm叫溶解温度(melting temperature, Tm),即是DNA双链溶解所需的温度。
大家可以理解,这个温度是由互补的DNA区域决定的,而不互补的区域对DNA的溶解是没有作用的。
因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。
计算Tm时,只计算互补的区域(除非你的酶切位点也与模板互补)。
不少战友设计的引物都Tm过低,是因为他们误把保护碱基和酶切位点都计算到Tm里了,最后的结果是导致了PCR反应的诸多困难。

构建重组质粒基本方法重组质粒是一种重要的遗传工程工具,用于将外源基因导入到宿主细胞中,从而实现特定基因的表达与功能研究。
构建重组质粒的基本方法可以概括为:选择质粒骨架、引物设计与合成、PCR扩增外源基因片段、DNA连接与重组、质粒扩增与提取、质粒鉴定与筛选,以下分别进行详细介绍。
一、选择质粒骨架在构建重组质粒时,首先需要选择一个合适的质粒骨架。
质粒骨架是指一个可复制的质粒DNA分子,常见的质粒骨架有pUC、pBR322、pET等。
质粒骨架上通常包含有宿主细胞可以识别的起始子和起始子附近的终止子,用于启动和终止转录过程,同时还包含选择标记基因,如抗生素抗性基因,以及其他在分子克隆中常用的诸如多克隆位点、限制酶切位点等。
二、引物设计与合成在构建重组质粒时,需要利用引物来扩增并克隆外源基因片段。
引物一般是两条DNA可控引物,其中一条是正向引物,另一条是反向引物。
引物的设计需要注意以下几点:引物的长度通常为15-30个碱基对,引物应该具有合适的Tm值,并且在引物双链的末端至少有2个碱基对是纯G或纯C。
引物可以使用商业引物合成公司合成。
三、PCR扩增外源基因片段使用引物扩增外源基因片段是构建重组质粒的一个关键步骤。
PCR反应一般包括DNA模板、引物、dNTPs和DNA聚合酶。
根据需要,可以使用特异性引物对目标基因进行PCR扩增,然后通过凝胶电泳检查PCR产物长度和纯度,并使用PCR产物进行下一步处理。
四、DNA连接与重组将PCR扩增得到的外源基因片段与质粒骨架进行连接和重组。
连接通常通过使用限制酶切和连接酶来实现。
限制酶切是利用限制酶切剪切DNA,生成具有互补粘性末端的DNA片段,然后将外源基因片段与质粒骨架进行连接。
连接酶可以使DNA片段之间的末端骨架参与phosphodiester结合反应,从而形成连体分子。
五、质粒扩增与提取将重组质粒转化到宿主细胞中,通过培养和培养基筛选来扩增质粒。
质粒扩增一般在含有抗生素的琼脂糖平板上进行,抗生素可以选择对宿主细胞有毒作用但不对重组质粒有毒的抗生素。

载体构建质粒构建步骤有哪些?载体构建过程:1、引物设计2、⽬的⽚段选取:RNA提取、RNA反转录、PCR扩增、PCR产物纯化3、双酶切4、连接:T4 DNA ligase连接或者同源重组连接(新贝⽣物:#B101、#B102)5、转化6、菌落PCR7、测序:1)摇菌;2)送样; 3)⽐对;8、菌种保存:菌种⽐对成功,则可保存菌种备⽤。
9、质粒提取:菌种⽐对成功,冻存菌种后,菌液⽤于提取质粒。
⼀、载体构建基本原理分、切、连、转、筛1、分:分离出要克隆的⽬的基因及载体。
2、切:⽤限制性内切酶切割⽬的基因和载体,使其产⽣便于连接的末端。
限制性内切酶:是⼀类能识别双链DNA中特定碱基顺序的核酸⽔解酶。
限制性核酸内切酶根据识别切割特性,催化条件及是否具有修饰酶活性分为三⼤类。
其中Ⅱ型酶能识别双链DNA的特异顺序,并在这个顺序内切割,产⽣特异性DNA⽚段,是DNA 重组技术中常⽤的酶。
Ⅰ型酶:具有修饰和切割功能,⽆固定切割位点Ⅲ型酶与Ⅰ型类似,能识别特异位点,但切割位点在识别位点以外Ⅱ型酶特点:①识别顺序⼀般为4-6个碱基对②识别顺序具有180度的旋转对称性,呈完全的回⽂结构③Ⅱ型酶对双链DNA两条链同时切割,可产⽣两种不同末端:平末端,粘末端平末端:在识别顺序的对称轴上,对DNA同时切割形成平末端,如:SmaI5’-CCC GGG-3’ 5’-CCC GGG-3’3’-GGG CCC-5’ 3’-GGG CCC-5’5′突出粘末端:在识别序列的两侧末端切割DNA双链,于对称轴的5 ′末端切割产⽣5 ′端突出的粘性末端,如:Hind Ⅲ5’―AAGCTT―3’ 5’― A 5’-AGCTT―3’3’―TTCGAA―5’ 3’― TTCGA-5’ A―5’3′突出粘末端:与5′突出粘末端作⽤相反,产⽣3 ′端突出粘末端,如:PstI5’―CTGCAG―3’ 5’―CTGCA-3’ G―3’3’―GACGTC―5’ 3’―G 3’-ACGTC―5’3、连:将切割后的⽬的基因和载体⽤T4 DNA连接酶连接或者同源重组⽅法连接。

DNA(质粒和PCR产物)测序操作程序一、P CR反应体系标准反应体系(20 μL)试剂用量DNA X μL(根据模板确定)BigDye 8 μL引物 (3.2 pmol / μL) 1 μL灭菌去离子水补充到20 μL总体积20 μL优化反应体系(10 μL)试剂用量DNA X μL (根据模板确定)BigDye 2 μL引物 (3.2 pmol / μL) 1 μL灭菌去离子水补充到20 μL总体积10 μL测序PCR热循环条件:96 ︒C 1 min96 ︒C 10 se50 ︒C 5 sec x 25个循环60 ︒C 4 min4 ︒C保温二、测序产物纯化:10 μL反应体系,384孔板,酒精/EDTA/NaAc法1.每管加入1 μL 125 mM EDTA到管底,每管加入1 μL 3M NaAc到管底。
2.每管加入25 μL 100%酒精,铝箔封严密,震荡混匀4次,室温放置15 min。
3.1400-2000 x g离心45 min 或者2000-3000 x g离心30 min,马上倒置384孔板,离心至185 x g 停止离心(从离心机启动到达到185 x g停止离心总共1 min时间)。
4.每管加入35 μL 70% 酒精,1650 x g 4 ︒C离心15 min;马上倒置384孔板,离心至185 xg 停止离心(从离心机启动到达到185 x g停止离心总共1 min时间)。
5.重复第4步1次。
6.室温挥发净酒精,加入10 μL Hi-Di Formamide溶解DNA;或者铝箔密封后于4 ︒C保存。
7.溶解后的样品需要在 95 ︒C变性4 min,迅速置冰中冷却4 min后,上样电泳。
三、测序产物纯化:单离心管法请参照20 μL反应体系,96孔板,酒精/EDTA/NaAc法操作,只需要将“倒置96孔板,离心至185 x g 停止离心”这一操作改为用枪吸尽上清液。
1.每管加入2 μL 125 mM EDTA到管底,每管加入2 μL 3M NaAc到管底。

质粒测序引物设计
质粒测序是一种常用的分子生物学技术,用于分析质粒的基因组成和结构。
在质粒测序过程中,引物是至关重要的组成部分,其质量和设计直接影响到测序数据的准确性和可靠性。
因此,正确设计合适的引物至关重要。
设计引物的首要步骤是选择目标序列。
质粒测序通常需要分析全长或部分序列,因此需要确定目标序列的起止位置。
在选择目标序列时,还需要考虑序列的特点,如长度、GC含量和反向互补性等。
这些因素将影响引物的长度和特异性。
设计引物时,需要遵循一些基本原则。
首先,引物的长度应该在18-25个碱基对之间,以保证特异性和特异性。
其次,引物的GC含量应该在40-60%之间,以确保引物与目标序列的结合。
此外,还需要确保引物的特异性,以避免与其他序列的杂交信号。
引物设计时还需要考虑引物间的距离和方向。
引物之间的距离应该足够远,以避免重叠或杂交。
另外,正向和反向引物之间的距离应该相等,以确保扩增产物的长度均匀。
总之,正确设计引物对质粒测序的结果至关重要。
根据目标序列的特点,采用合适的引物设计策略和软件,可以设计出高质量的引物,从而获得更加准确和可靠的测序数据。
- 1 -。
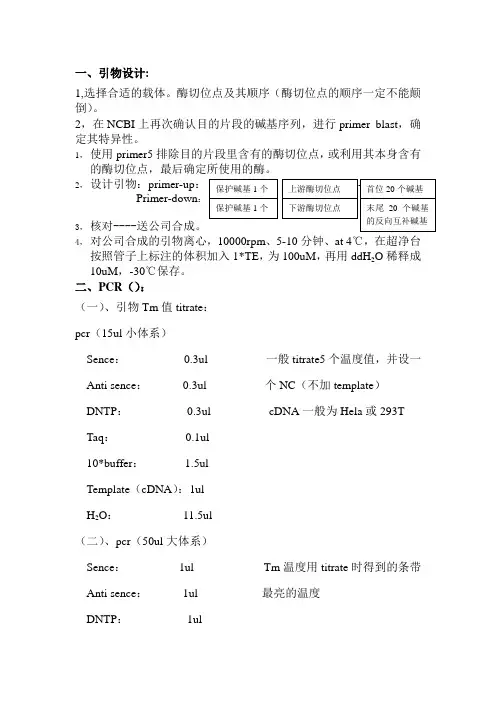
一、引物设计:1,选择合适的载体。
酶切位点及其顺序(酶切位点的顺序一定不能颠倒)。
2,在NCBI上再次确认目的片段的碱基序列,进行primer blast,确定其特异性。
1,使用primer5排除目的片段里含有的酶切位点,或利用其本身含有的酶切位点,最后确定所使用的酶。
2,设计引物:primer-up:Primer-down:3,核对----送公司合成。
4,对公司合成的引物离心,10000rpm、5-10分钟、at 4℃,在超净台按照管子上标注的体积加入1*TE,为100uM,再用ddH2O稀释成10uM,-30℃保存。
二、PCR():(一)、引物Tm值titrate:pcr(15ul小体系)Sence:0.3ul 一般titrate5个温度值,并设一Anti sence:0.3ul 个NC(不加template)DNTP:0.3ul cDNA一般为Hela或293T Taq:0.1ul10*buffer: 1.5ulTemplate(cDNA):1ulH2O:11.5ul(二)、pcr(50ul大体系)Sence:1ul Tm温度用titrate时得到的条带Anti sence:1ul 最亮的温度DNTP:1ulTaq:0.5ul 10*buffer:5ul Template(cDNA):2ul H2O:39.5ul三、跑胶,胶回收:称1.2g的琼脂糖加入100ml的plasmid越小胶浓度越大)胶凝固后,即可点样跑胶。
2、跑胶:120V、40min。
3、泡EB溶液20min后,紫外灯下观察,切胶(要带防护手套和口罩)4、做胶回收:(1)加入400ul溶胶液,55℃溶胶;(2)待胶全溶后,过胶回收柱,12000rpm,1min;(3)DNA Wash Buffer 500ul,12000rpm,1min,twice;(4)开盖离心,15min;(5)换新的1.5ml EP管,加Elution Buffer 30ul,12000rpm,1min,重复吸取,12000rpm,1min;(Elution Buffer使用前65℃温热)(6)测浓度四、连接:2x Buffer 5ul10ul的体系T Vector 0.5ul 16℃水浴过夜Insert 4ulLignase 0.5ul若insert浓度较大,应加的量小于4ul,则其余部分用水补齐。

(lnc2为例,lnc2分三段连接,GTP1、GTP2、GTP3(具体见文档“lncRNA4构建质粒的基本步骤:和lncRNA2的实验说明”中lnc2的部分))基本流程:设计三个片段的引物(设计时注意载体pcDNA3.1的酶切位点)普通PCR 琼脂糖凝胶电泳后胶回收酶切电泳后胶回收酶连转化涂板挑单菌落(10管)摇菌提质粒酶切初步鉴定测序具体步骤:1、设计引物2、先将GTP1进行普通PCR(三个片段的模板分别为单独连接有该片段的质粒),扩增条件聚合酶的说明书上有,50μl体系。
3、制胶(琼脂糖凝胶),将PCR产物全部上样,电泳(电泳液要新配置)45min左右后放紫外灯下,用干净刀片切出目的片段放置干净EP管,紫外灯每次照射时间不能太长。
4、用胶回收试剂盒回收,具体步骤见说明书。
5、胶回收后,用Nhe I,Kpn I两种酶将胶回收产物和载体pcDNA3.1进行双酶切5小时。
6、制胶,将酶切产物全部上样,跑电泳,胶回收(同步骤3、4)7、配酶连体系(见文档“分子生物学反应体系”),反应条件见连接酶说明书。
一般4℃过夜8、转化,(具体步骤见文档“分子生物学反应体系”)9、挑单菌落,10管,在加有氨苄的LB的试管中培养12~16h,37℃,140rpm.10、小提质粒(具体步骤见质粒提取试剂盒说明书)11、根据片段设计相关酶进行酶切鉴定,刷选出目的质粒,送去测序12、GTP1连上去后,GTP2、GTP3也与上述方法一样进行操作,只是GTP2的载体变为pcDNA3.1-- GTP1,GTP3的载体变为pcDNA3.1-- GTP1-- GTP2,同时相关的双酶切的酶也不同,GTP2片段时用Kpn I,BamHI。
GTP3片段时用BamHI,Xho I。
质粒构建流程质粒构建是分子生物学实验中常见的一项重要技术,它可以用于基因克隆、蛋白表达、基因编辑等多个领域。
在本文中,我们将介绍质粒构建的基本流程,希望能够帮助大家更好地理解和掌握这一技术。
第一步,设计引物。
质粒构建的第一步是设计引物。
引物是一小段单链DNA,它们的序列与目标DNA的两端相互补。
在构建质粒时,我们需要设计两对引物,分别用于扩增目标DNA的两端。
引物设计的好坏将直接影响到后续的实验效果,因此需要仔细选择引物的序列,确保其具有高度的特异性和稳定性。
第二步,PCR扩增。
设计好引物后,接下来就是进行PCR扩增。
PCR是一种体外合成DNA的方法,通过PCR扩增可以在短时间内获得大量目标DNA。
在PCR反应中,我们需要将待扩增的DNA模板、引物、DNA聚合酶和反应缓冲液混合,然后进行一系列的温度循环,最终得到目标DNA的扩增产物。
第三步,酶切和连接。
获得PCR产物后,接下来需要进行酶切和连接。
酶切是利用限制性内切酶在特定的酶切位点上切割DNA分子,从而得到特定的DNA片段。
在质粒构建中,我们通常会选择两种不同的限制性内切酶,分别用于酶切目标DNA和质粒载体。
然后将酶切后的目标DNA 片段与质粒载体连接,形成重组质粒。
第四步,转化和筛选。
最后一步是将重组质粒转化至宿主细胞中,然后进行筛选。
转化是利用化学方法或电穿孔法将质粒导入宿主细胞内,使其在细胞内进行复制和表达。
然后通过对转化后的细胞进行抗生素筛选或荧光筛选,筛选出含有目标重组质粒的细胞克隆。
总结。
质粒构建是一项复杂而又重要的实验技术,它涉及到许多分子生物学的基本原理和实验操作。
通过本文的介绍,相信大家对质粒构建的流程有了更清晰的认识。
当然,质粒构建的具体操作还需要根据实验的具体要求和目的进行调整和优化。
希望本文能够为大家在质粒构建实验中提供一些帮助和指导。
学院:______ 班级:_______ 学号:_________ 姓名:__________ 成绩:______ 实验二引物设计及测序结果分析目的:1、掌握常规引物设计的原则及操作流程。
2、熟悉简并引物设计的原理及操作方法。
3、熟悉引物设计软件及在线引物设计工具的操作方法。
4、掌握使用相关软件及在线工具分析测序结果的方法。
内容:1、使用Primer Premier、Oligo、BLAST等软件及在线工具进行常规引物设计,并对引物扩增效率、特异性进行评价。
2、使用DNAMAN软件进行常规引物快速设计。
3、使用NCBI中的在线引物设计工具Primer-BLAST快速设计引物。
4、使用在线工具CODEHOP设计简并引物。
5、使用Chromas、BioEdit软件查阅测序结果峰图文件。
6、使用DNAMAN软件对测序序列进行编辑,进行序列拼接。
软硬件要求:联网计算机,预装Windows 7操作系统,预装IE或Chrome浏览器、英汉电子词典(有道词典或金山词霸),预装DNAMAN7、Primer Premier5、Oligo7、Chromas、BioEdit等生物信息学分析软件。
操作及问题:一、Primer Premier5、Oligo7、BLAST常规引物设计本部分操作将使用Primer Premier5、Oligo7、BLAST等软件及工具设计拟南芥AtBADH基因编码区全长特异引物。
(参考“第四章引物设计及测序结果分析”课件)(一)使用Primer Premier5搜索引物1、在NCBI数据中查找登录号为NM_001198470的序列记录,查阅相关信息,并下载序列将其保存为fasta格式文件。
问题1:该序列是什么类型的序列?该序列编码区在什么位置?2、打开Primer premier5软件,点击键ctrl+V将上一步中下载的序列粘贴入弹出的GeneTank窗口中(或者点击。
3、点击GeneTank窗口中左上角的Primer premier窗口中点击Search Criteria窗口中根据要求选择合适选项及参数,选定后,点击Search Progress窗口中有Search Results窗口;如没有出现数重新搜索引物。
质粒构建流程一、引物设计1)取得目的基因序列,可选用数据库NCBI2)软件分析目的基因可用酶切位点。
使用primer5分析出序列不包含的酶切位点,即为可用没切位点。
3)4)选择酶切位点。
对照目的基因可用酶切位点和载体上的酶切位点,选择二者共有的作为备选。
载体上两个酶切位点的距离应有几十bp以上,选实验室常用酶切位点。
5)使用primer5设计目的基因引物,目的产物应包含从启动子到终止子全部碱基。
6)根据选择的酶切位点,查找对应的酶切位点保护碱基,将对应片段添加到设计的引物两端,注意酶切位点的前后顺序。
一般选择三个保护碱基。
7)引物设计完成,送公司合成。
二、目的片段获取1. RNA提取试剂盒:Bioteke高纯总RNA快速提取试剂盒离心柱型(裂解液RL 4℃、漂洗液RW -20℃保存)准备:冰盒、4℃预冷离心机、EP管2套、吸附柱RA一套操作步骤:1)将1000 Hl裂解液RL加入细胞中,混合5min。
2)加200 Hl氯仿混合,震荡15$,室温孵育3min。
3)4℃, 12000rpm 离心 10min。
4)最上层水相转移至新EP管中(体积约550 H1)5)加入1倍体积(550 H1) 70%乙醇,混匀6)全部转移到套收集管的吸附柱RA中,4℃, 10000rpm离心45s7)弃废液,重套收集管,加500 Hl去蛋白液RE, 12000rpm离心45s8)弃废液,重套收集管,加700 H l去漂洗液RW, 12000rpm离心60s9)弃废液,重套收集管,加500 H l去漂洗液RW, 12000rpm离心60s10)弃废液,重套收集管,12000rpm空离2min11)吸附柱放入新EP管,加50 Hl RNase free water于膜上,室温放置2min12)4℃, 12000rpm 离心 60s13)点样:5Hl RNA+ 1 H l 10X14)-20℃保存2. RNA反转录试剂盒: TaKaRa primescript RT reagent kit with gDNA eraser (-20℃保存)准备:冰盒,②④⑤⑥取出解冻,①③为酶不可取出,预冷离心管操作步骤:1)基因组DNA去除(10Hl体系)② 5XgDNA eraser buffer 2Hl① gDNA eraser 1HlTotal RNA 4Hl (可根据RNA浓度调整)⑥ RNase free water 3HlPCR仪中进行,程序:42℃, 2min f4℃注:RNA的量可根据浓度调整,混合液冰上配制,酶最后加入2)反转录反应(20Hl体系)④5XprimerScript buffer 2 4ul⑤primerScript RT enzyme mix I 1ul⑥RT primer mix 1ul⑦RNase free water 4ul1)反应液10 HlPCR 仪:37℃, 15min—85℃, 5s―4℃注:可直接将第2步反混合液好后加入到第1步反应液中-20℃长期保存3.PCR扩增高保真酶primerstar扩增,50 Hl体系如下:5XPS buffer 10Hl PCR程序:dNTP 4Hl 95℃5minHl 95℃30sHl 56℃30s , 35cyclecDNA 1 H l(可变)72℃1minR-primer 1Hl 72℃10minF-primer 1 H l 注:可根据不同基因调节退货温度,延伸时点样:5Hl PCR 产物+ iHl 6X4.PCR产物纯化1)液相纯化(产物电泳结果只含目的条带)试剂盒:Microelute cycle-pure spin protocol(OMEGA bio-tek) D6293-01①②转移至HiBind MicroElution DNA柱(套收集管),室温离心10000g, imin。
检测质粒polya尾长度的引物和方法与流程质粒是一种常见的DNA分子,在分子生物学研究中起着至关重要的作用。
质粒上的多聚腺苷酸酸尾(Poly(A) tail)是由若干个腺嘌呤碱基(A)组成的一段聚合物。
检测质粒Poly(A)尾的长度可以帮助我们了解质粒的稳定性和表达效率,并对下游实验设计提供指导。
本文将介绍检测质粒Poly(A)尾长度的引物、方法和流程。
一、引物选择:检测质粒Poly(A)尾长度的引物选择关键是针对Poly(A)序列进行扩增。
常用的引物有两个:Oligo(dT)引物和Random Hexamer引物。
1. Oligo(dT)引物:Oligo(dT)引物是一种寡聚腺苷酸引物,它具有高特异性,只能与Poly(A)序列发生特异性的碱基互补配对。
因此,Oligo(dT)引物非常适合用于检测Poly(A)尾。
2. Random Hexamer引物:Random Hexamer引物是一种随机引物,它由六个随机碱基序列组成。
Random Hexamer引物可以结合在RNA的任意位置,起到随机引物的作用。
然而,由于Poly(A)尾只有腺苷酸组成,所以Random Hexamer引物在检测Poly(A)尾长度时需要配合使用适当的控制样本。
二、检测质粒Poly(A)尾长度的方法和流程:检测质粒Poly(A)尾长度的方法和流程通常包括以下几个步骤:RNA提取、逆转录反应、PCR扩增和电泳分析。
1. RNA提取:首先,需要提取目标质粒中的RNA。
RNA提取应该选择合适的试剂盒,根据厂商提供的实验方案进行操作。
常见的RNA提取试剂盒有TRIzol试剂盒、RNAeasy试剂盒等。
2.逆转录反应:RNA提取后,需要进行逆转录反应,将RNA反转录成cDNA。
逆转录反应时需要使用逆转录酶和引物,常用的逆转录酶有M-MLV逆转录酶和Superscript逆转录酶。
在逆转录反应中,可以选择使用Oligo(dT)引物或者Random Hexamer引物。
质粒 pcr传代稳定性引物设计流程或方法下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!质粒 PCR 传代稳定性引物设计流程或方法质粒是细菌和古细菌细胞中常见的环状 DNA 分子,广泛应用于基因工程实验和生产中。
质粒测序引物设计
随着基因研究的不断深入,质粒测序成为了分析基因信息的重要工具之一,同时,为了获得更真实、更准确的序列,准确设计序列特异性强的引物是非常重要的。
本文将重点讲述质粒测序所需的引物设计流程及其注意事项,以期为大家提供参考。
1. 引物设计的基本原则引物是一种在PCR反应体系中使DNA聚合酶特异性扩增所需的寡核苷酸。
正确认识引物的作用和特点,对于准确设计合适的引物具有很重要的意义。
引物的主要特点有以下几个方面:
(1)序列特异性:引物应特异性地与目标序列结合,确保扩增出的片段为所需的序列,而非与其它非靶DNA结合。
(2)稳定性:使其与模板DNA的双链DNA进行稳定的配对。
(3)温度特异性:引物与模板结合的温度应该略低于生产它们的反应体系的温度。
基于以上特点,我们需要基于引物设计的原则进行质粒测序引物的设计。
具体来说,主要考虑以下几个方面:(1)序列特异性:引物应设计成特异性的,避免产生与目标序列无关的PCR产物。
其特异性应通过序列比对和BLAST进行验证,以确保最终设计出的引物能够对所需的目
标序列特异性扩增。
在引物设计时,还应注意要在引物中避开GC/AT区域和重复序列的部位。
(2)长度优化:引物的长度应该合适,过短容易导致扩增不全,过长则容易出现引物间的竞争关系,同时也容易产生温度特异性方面的问题。
一般来说,引物长度应该控制在20个核苷酸左右,但是对于某些特殊的情况,比如复杂基因组、高度变异基因等,可能需要设计更长的引物。
(3)序列特征:引物中不应含有重复结构、长程序列、寡聚物或者常见的RNA序列,这些都可能导致不特异扩增、引物二聚或聚合酶滞留等问题。
2. 引物设计流程
在对质粒进行测序之前,需要先设计合适的引物,紧密契合上述引物设计原则,具体流程如下:
(1)数据获取:首先需要获取目标DNA序列,并确定所需要进行的PCR扩增区域。
(2)引物设计:根据目标区域,按照上述设计原则进行引物的设计。
一般来说,可以利用一些公共的软件或者在线工具进行引物设计,比如Primer 3、OligoCalc、Primer Premier等。
(3)引物评估:设计好的引物需要进行验证,包括序列特异性、设计可靠性等方面。
可以利用序列比对、
BLAST、熔解曲线分析、三维结构分析等手段进行引物的评估。
如果存在不确定因素,还需要进行实验验证,可以利用端点PCR、荧光比色PCR、分子条带电泳等方法进行验证。
(4)引物改进:如果引物设计后出现问题,如存在非特异扩增产物、引物JC值过高等情况,可以通过引物的结构调整、长度变化、位置调整等手段进行引物的改进。
3. 引物设计注意事项
引物设计在实际应用中也存在一些值得注意的问题,具体来说主要有以下几个方面:
(1)检查引物特异性:在引物设计过程中,我们需要对每个引物的特异性进行检查,确保设计的引物可以特异性扩增所需的目标序列。
(2)定量引物浓度:在实际操作中,我们需要按照一定的比例来定量引物浓度。
如果引物的浓度过低,则可能导致扩增效率不高,产物浓度不够;如果引物的浓度过高,则可能出现引物之间的竞争关系,甚至会出现非特异性扩增产物。
(3)注意引物之间的互相作用:引物在反应体系中会相互作用,如果引物之间的互相作用过强,那么就可能会导致非特异性扩增产物产生,这时候需要调整引物浓度的配比。
总之,在进行质粒测序的过程中,引物的设计是一个比较关键的步骤,合理、科学的引物设计可以获得更加稳定、可靠的实验结果,对于我们理解基因信息具有很大的帮助。