替诺福韦酯的产业化生产工艺
- 格式:doc
- 大小:137.00 KB
- 文档页数:7

替诺福韦的合成工艺改进_刘嘉(优选)word资料论著替诺福韦的合成工艺改进刘嘉 , 李科 , 孙海玲 , 冯继禄 (第二军医大学药学院药物化学教研室 , 上海200433作者简介 :刘嘉 (1980 , 男 , 硕士研究生 . E m ai:l nudt_li u ji a @hot m ai. l co m :l @si na . m摘要目的 :改进替诺福韦的合成工艺。
方法 :以亚磷酸二乙酯和多聚甲醛经缩合、酯化得到对甲苯磺酸羟甲基磷酸二乙基酯 (4 。
另以缩水甘油合成碳酸丙烯酯 , 与腺嘌呤反应得到 9 (2 羟丙基腺嘌呤 (7, 在叔丁醇钠作用下与 (4 经缩合、水解等反应制得抗病毒药物替诺福韦。
结果 :优化后的制备工艺成本低 , 操作简便 , 对环境污染减少。
结论 :新工艺的产率达到 30%, 适合工业生产。
关键词替诺福韦 ; 合成 ; 工艺改进中图分类号 :R914 文献标识码 :A 文章编号 :1006-0111(2021 01-0031-03I mprove m ent of synthetic process of tenofovirL I U Ji a , L I K e , S UN H ai ling , FENG Ji l u (D epart m ent o fM ed ici na l Chem istry , Schoo l of phar m acy , SecondM ilitary M ed i ca lU n i versity , Shangha i 200433, Ch i naAB STRACT O b jective :T o i m prove the synthe ti c process of teno f ov ir . M eth ods :P t o luenes u lfonic ac i d diethoxyphosphoryl me t hy l ester(4 w as prepared from d i ethy l phosphate and parafor m a l dehyde by condensa tion and este rifica ti on .9 (2 hydroxy propy l aden i ne (7 w as synt hesized by reacti on o f aden i ne and propy lene ca rbonate . In presence o f (C H 3 3CON a , co m pound (7 was connected w ith compound (4, and then hydro l y zed to teno fov ir . R esu lts :T he technique w as s uccessfull y i m proved due t o its l ow co st , easi e r opera ti on and less po ll u ti on tothe env iron m en t . Conclusion :The overall y ield o f t he i m proved synthetic process was 30%, wh ich is m ore suitable fo r i ndustria l producti on . K EY W ORDStenofov ir ; synthesis ; techn ica l i m prove m ent替诺福韦酯 (teno fov ir d isoprox il fum ara te , 替诺福韦双异丙酰氧基甲酯富马酸盐 , 化学名为 (R [[2 (6 氨基 9H 嘌呤 9 基 ] 1 甲基乙氧基 ]甲基 ]膦酸双(异丙酰氧基甲基酯富马酸盐 , 是一可口服的开环核苷酸单膦酸酯的前药 , 结构式如图 1(1 所示 , 口服吸收后迅速转变成替诺福韦 (P M P A, 2[1]。
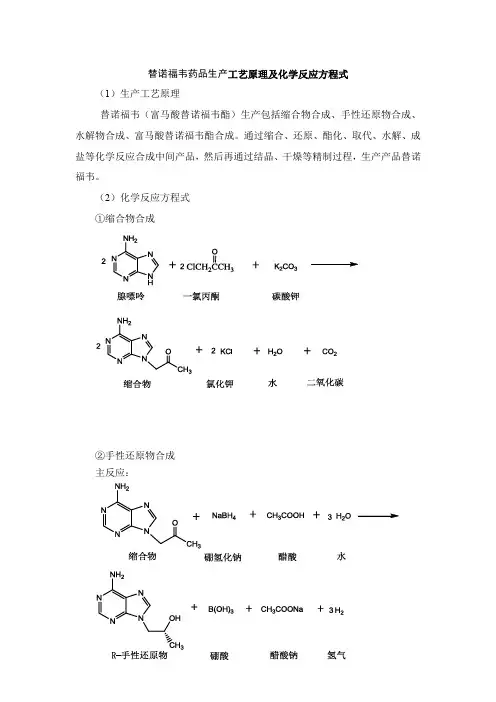
替诺福韦药品生产工艺原理及化学反应方程式
(1)生产工艺原理
替诺福韦(富马酸替诺福韦酯)生产包括缩合物合成、手性还原物合成、水解物合成、富马酸替诺福韦酯合成。
通过缩合、还原、酯化、取代、水解、成盐等化学反应合成中间产品,然后再通过结晶、干燥等精制过程,生产产品替诺福韦。
(2)化学反应方程式
①缩合物合成
②手性还原物合成
主反应:
副反应:
③水解物合成
A.DESMP合成
主反应:
DESMP全称:对甲苯磺酰氧甲基膦酸二乙酯副反应:
B.磷酸酯合成
主反应:
副反应:
C.水解物合成主反应:
副反应:
④富马酸替诺福韦酯合成
A.替诺福韦酯合成
B.替诺福韦酯富马酸盐合成。

抗病毒药物替诺福韦(Tenofovir)的最新合成工艺解析本文主要介绍氧代甲基膦酸二叔丁酯用于活性药物成分替诺福韦(Tenofovir,PMPA)合成的相关研究。
首先,开发了一种简单且有效的合成二叔丁基(羟甲基)膦酸酯的方法。
同时,通过O-甲磺酰化以高收率生成氧代甲基膦酸二叔丁酯。
随后,使用Mg(O t Bu)2作为碱,通过两步法实现(R)-9-(2-羟丙基)腺嘌呤(HPA,4)和13的烷基化以及脱保护反应,从而合成PMPA,收率为68%。
若进行5g实验,可将收率提高至72%。
此外,该策略也可用于乙型肝炎药物阿德福韦(Adefovir)的合成,收率为64%。
正文:1993年,Balzarini报道了核苷酸逆转录酶抑制剂替诺福韦(1,PMPA),用于人类免疫缺陷病毒(HIV)药物。
为实现口服应用,并增加生物利用度,从而开发出TDF(2),于2001年被FDA批准用于HIV治疗,并于2008年用于乙肝病毒的治疗。
2010年,又开发一种TAF(3),与TDF相比,具有更少的副作用和更好的耐受性(Figure 1)。
此外,在2020年COVID-19大流行期间,替诺福韦作为治疗SARS-CoV2的潜在药物,并用于各项研究。
2010年,Ripin等报道了一种(R)-9-(2-羟丙基)腺嘌呤(HPA,4)和DESMP(5)的烷基化以及脱保护反应,其中使用Mg(O t Bu)2作为碱时,转化率> 90%。
虽然该反应使用TMCSI/NaBr代替昂贵的TMSBr(降低脱保护的成本),但后处理操作复杂,需多次过滤和萃取,且滤饼中损失高达15%等。
最终,以两步59%的总收率获得PMPA·H2O。
2016年,Riley等报道了通过MeMgCl和t BuOH原位生成Mg(O t Bu)2,并对脱保护(HBr/乙酸)工艺进行改进,最终以两步57%收率获得PMPA·H2O。
2020年,Derstine等报道了一种以NaO t Bu作为碱,于DMF溶剂中实现HPA与7的烷基化反应,从而获得70%收率的PMPA。
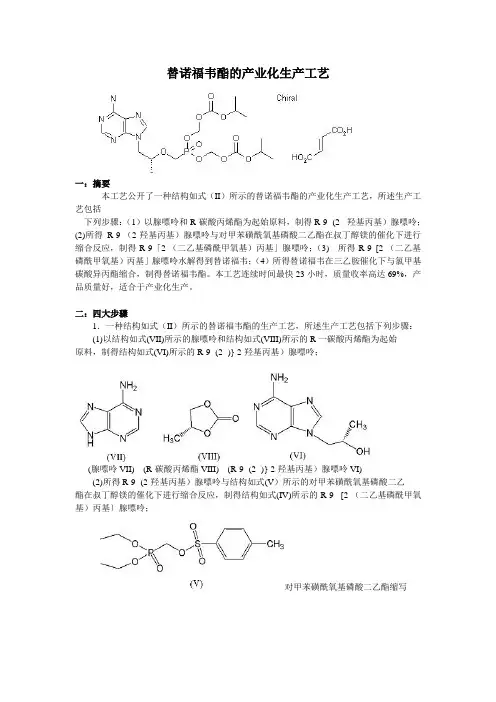
替诺福韦酯的产业化生产工艺一:摘要本工艺公开了一种结构如式(II)所示的替诺福韦酯的产业化生产工艺,所述生产工艺包括下列步骤:(1)以腺嘌呤和R-碳酸丙烯酯为起始原料,制得R-9- (2- 羟基丙基)腺嘌呤;(2)所得R-9-(2-羟基丙基)腺嘌呤与对甲苯磺酰氧基磷酸二乙酯在叔丁醇镁的催化下进行缩合反应,制得R-9「2-(二乙基磷酰甲氧基)丙基」腺嘌呤;(3) 所得R-9 [2-(二乙基磷酰甲氧基)丙基」腺嘌呤水解得到替诺福韦;(4)所得替诺福韦在三乙胺催化下与氯甲基碳酸异丙酯缩合,制得替诺福韦酯。
本工艺连续时间最快23小时,质量收率高达69%,产品质量好,适合于产业化生产。
二:四大步骤1.一种结构如式(II)所示的替诺福韦酯的生产工艺,所述生产工艺包括下列步骤:(1)以结构如式(VII)所示的腺嘌呤和结构如式(VIII)所示的R一碳酸丙烯酯为起始原料,制得结构如式(VI)所示的R-9- (2- )} 2-羟基丙基)腺嘌呤;(腺嘌呤VII) (R-碳酸丙烯酯VIII) (R-9- (2- )} 2-羟基丙基)腺嘌呤VI)(2)所得R-9- (2-羟基丙基)腺嘌呤与结构如式(V)所示的对甲苯磺酰氧基磷酸二乙酯在叔丁醇镁的催化下进行缩合反应,制得结构如式(IV)所示的R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤;对甲苯磺酰氧基磷酸二乙酯缩写DESMP(V) (IV)(3)所得R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤水解得到式(III)所示的替诺福韦;(III)(4)所得替诺福韦在弱碱性物质催化下与氯甲基碳酸异丙酯缩合,制得结构如式(II)所示的替诺福韦酯;1.1步骤(1)所述的替诺福韦酯的生产工艺,其特征在于所述步骤(1)具体包括:腺嘌呤和R一碳酸丙烯酯在碱性催化剂下缩合反应,充分反应后分离得到粗品,粗品经精制得到R-9-(2-羟基丙基)腺嘌呤。
1.2所述的R-9- (2-羟基丙基)腺嘌呤的生产工艺,其特征在于所述步骤(l)中:在充N2保护下,R一碳酸丙烯酯和腺嘌呤的摩尔比例为1. 25:1。

替诺福韦酯工艺流程
替诺福韦酯工艺流程如下:
1.替诺福韦酯合成。
氮气保护下,向反应釜中拉入1-甲基-2-吡咯烷酮,加入上步水解物和二异丙基乙胺,搅拌,升温至60℃缓慢滴加氯甲基碳酸异丙酯,滴完后保温至中控检测原料基本反应完全。
反应结束,减压蒸馏回收N-甲基吡咯烷酮套用,剩余物加入异丙醚重结晶,离心,固体在50℃真空干燥得中间产物。
2.富马酸替诺福韦酯合成。
将富马酸溶于异丙醇,搅拌下加入上步替诺福韦酯,升温至60℃左右反应6小时。
反应结束,冷却至0℃左右,搅拌5小时冷却析晶,离心,固体干燥、粉碎、包装得产物。
替诺福韦酯的产业化生产工艺一:摘要本工艺公开了一种结构如式(II)所示的替诺福韦酯的产业化生产工艺,所述生产工艺包括下列步骤:(1)以腺嘌呤和R-碳酸丙烯酯为起始原料,制得R-9- (2- 羟基丙基)腺嘌呤;(2)所得R-9-(2-羟基丙基)腺嘌呤与对甲苯磺酰氧基磷酸二乙酯在叔丁醇镁的催化下进行缩合反应,制得R-9「2-(二乙基磷酰甲氧基)丙基」腺嘌呤;(3) 所得R-9 [2-(二乙基磷酰甲氧基)丙基」腺嘌呤水解得到替诺福韦;(4)所得替诺福韦在三乙胺催化下与氯甲基碳酸异丙酯缩合,制得替诺福韦酯。
本工艺连续时间最快23小时,质量收率高达69%,产品质量好,适合于产业化生产。
二:四大步骤1.一种结构如式(II)所示的替诺福韦酯的生产工艺,所述生产工艺包括下列步骤:(1)以结构如式(VII)所示的腺嘌呤和结构如式(VIII)所示的R一碳酸丙烯酯为起始原料,制得结构如式(VI)所示的R-9- (2- )} 2-羟基丙基)腺嘌呤;(腺嘌呤VII) (R-碳酸丙烯酯VIII) (R-9- (2- )} 2-羟基丙基)腺嘌呤VI)(2)所得R-9- (2-羟基丙基)腺嘌呤与结构如式(V)所示的对甲苯磺酰氧基磷酸二乙酯在叔丁醇镁的催化下进行缩合反应,制得结构如式(IV)所示的R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤;对甲苯磺酰氧基磷酸二乙酯缩写DESMP(V) (IV)(3)所得R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤水解得到式(III)所示的替诺福韦;(III)(4)所得替诺福韦在弱碱性物质催化下与氯甲基碳酸异丙酯缩合,制得结构如式(II)所示的替诺福韦酯;1.1步骤(1)所述的替诺福韦酯的生产工艺,其特征在于所述步骤(1)具体包括:腺嘌呤和R一碳酸丙烯酯在碱性催化剂下缩合反应,充分反应后分离得到粗品,粗品经精制得到R-9-(2-羟基丙基)腺嘌呤。
1.2所述的R-9- (2-羟基丙基)腺嘌呤的生产工艺,其特征在于所述步骤(l)中:在充N2保护下,R一碳酸丙烯酯和腺嘌呤的摩尔比例为1. 25:1。
腺嘌呤和碱性催化剂的摩尔比例为10:1;反应以二甲基甲酰胺(DMF)或二甲基亚砜(DMSO)为反应溶剂,所述的碱性催化剂为氢氧化钠或氢氧化钾;反应温度控制在100一130℃;具体包括:腺嘌呤和R一碳酸丙烯酯在碱性催化剂下缩合反应,充分反应10hrs后,用HPLC监控反应的进行,通过原料含量的变化确定反应终点。
将反应物母液分离得到含有R-9- (2- )} 2-羟基丙基)腺嘌呤的粗品,粗品精制采用的精制溶剂为甲醇或乙醇和甲苯的混合物,其中甲苯在混合物中的体积含量不超过30%(乙醇:甲苯=75:25)。
粗品经精制得到R-9- (2-羟基丙基)腺嘌呤。
2.1步骤(2)具体包括:无水无氧条件下,R-9-(2-羟基丙基)腺嘌呤在叔丁醇镁的催化下与对甲苯磺酰氧基磷酸二乙酯缩合;充分反应后用有机酸中和,蒸除溶剂,加入二氯甲烷和水,搅拌后层析出镁盐,过滤分水,有机层浓缩得到R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤。
由于反应以叔丁醇镁为催化剂,大量的镁离子会带入到后续反应中,导致产品杂质大大提高,也影响收率。
考虑到有机酸的镁盐水溶性较大,所以我们采用有机酸直接中和镁离子,在此中除去镁盐。
中和所用的有机酸优选为甲酸、乙酸或丙酸,更优选为甲酸。
2.2所述的R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤的生产工艺,其特征在于所述步骤(2)中:充N2,R-9- (2-羟基丙基)腺嘌呤与脱水干燥过的对甲苯磺酰氧基磷酸二乙酯的缩合反应,以二甲基甲酰胺为反应溶剂;所述R-9- (2-羟基丙基)腺嘌呤与对甲苯磺酰氧基磷酸二乙酯的投料摩尔比为1:1. 5;R-9- (2-羟基丙基)腺嘌呤和催化剂叔丁醇镁的投料摩尔比为1:1.1一1:1.2;R-9- (2-羟基丙基)腺嘌呤与对甲苯磺酰氧基磷酸二乙酯的缩合反应温度为60一90℃;:在无水无氧条件下,R-9-(2-羟基丙基)腺嘌呤在叔丁醇镁的催化下与对甲苯磺酰氧基磷酸二乙酯缩合,充分反应2.5hr后,用HPLC监控反应的进行,通过原料含量的变化确定反应终点。
用有机酸中和PH=7,加热蒸除溶剂回收,加入二氯甲烷和水,搅拌后析出镁盐,过滤分离水层,有机层浓缩得到R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤。
3.1所述的替诺福韦的生产工艺,其特征在于步骤(3)所述的水解优选在水解试剂氢溴酸的作用下进行,反应快速,原料价廉易得。
所述的步骤(3)具体包括:将步骤(2)所得R-9-「2-(二乙基磷酰甲氧基)丙基〕腺嘌呤加入氢溴酸水溶液中,升温水解,充分反应后冷却,加入有机溶剂萃取,水层用碱液调节pH值为3.0-3.3,冷却析晶,过滤,水精制,干燥后得到替诺福韦。
3.2 所述的替诺福韦的生产工艺,其特征在于所述步骤中:萃取(3)用有机溶剂选自下列之一:二氯甲烷、乙酸乙酯、正己烷、氯仿;水解反应温度控制在80-100℃;所述R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤与氢溴酸的投料摩尔比为1:7-1:7.5。
:将步骤(2)所得R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤加入氢溴酸水溶液中,升温水解,充分反应6hr后,用HPLC监控反应的进行,通过产生副产物单乙酯含量的变化确定反应终点。
冷却反应液,加入有机溶剂中萃取,水层用碱液调节pH值为3.0一3. 3,冷却析晶,过滤,水精制,干燥后得到替诺福韦。
3.3 以上(3)中,水解反应温度优选95℃。
为提高产品纯度,加入有机溶剂萃取以除去未反应完全的原料和生成的单乙酯(即只脱去一个乙基的副产品)。
萃取用有机溶剂优选为二氯甲烷或乙酸乙酯。
4.1(4)所述的替诺福韦酯的生产工艺,其特征在于所述步骤(4)具体包括:替诺福韦和氯甲基碳酸异丙酯在弱碱性物质的催化下缩合,充分反应后冷却,加入水和二氯甲烷萃取,碱性水溶液洗涤二氯甲烷层,干燥、蒸除二氯甲烷得到粗品,粗品中加入低极性溶剂,冷却析晶得到替诺福韦酯。
4.2所述的替诺福韦酯的生产工艺,其特征在于所述步骤(4)中:所述弱碱性物质为三乙胺、碳酸氢钠或碳酸钠混合水溶液。
所述替诺福韦和氯甲基碳酸异丙酯的反应以1一甲基一2一吡咯烷酮为反应溶剂,替诺福韦和氯甲基碳酸异丙酯的投料摩尔比优选为1:5,反应温度控制在55℃,洗涤二氯甲烷层所采用的碱性水溶液的pH=8.0,所述低极性溶剂选自下列之一:乙酸乙酯、环己烷、石油醚、正己烷。
具体包括:替诺福韦和氯甲基碳酸异丙酯在弱碱性物质的催化下缩合,充分反应4hrs后,用HPLC监控反应的进行,当单(POC) 酯含量为20%以下为最佳停止点。
冷却反应液,加入水和二氯甲烷萃取,最终产物中,单(POC) 酯的杂质非常容易超标,并且很难通过精制的方法直接除去,所以我们采用碱性水溶液洗涤有机层以除去产品中的副产物单酯,优选碱性水溶液的pH在7.5-8,若碱性太强容易导致产品分解并且消旋化。
所述碱性水溶液可选用pH在7. 5一10.0的三乙胺、碳酸氢钠、碳酸钠等水溶液。
用碱性水溶液洗涤二氯甲烷层,干燥、蒸除/回收二氯甲烷得到替诺福韦酯粗品,粗品中加入低极性溶剂石油醚,冷却析晶后干燥1hr,得到替诺福韦酯。
三:工艺概括[1]本工艺第一步骤产率73%;第二步骤产率95%;第三步骤产率55%;第四步骤产率55%;原子总利用率21%,。
[2]本工艺所述生产工艺操作简便,所有的原材料都价廉和易得,此工艺最大的优点主要体现在如下方面:[3] 没有采用正丁基锂或者钠氢这些催化剂的使用,提高了生产的安全性;此工艺提供了一种高纯度的富马酸替诺福韦酯的生产工艺,解决了副产物单乙酯、单(POC) 酯以及手性异构体杂质在产品中含量过高的问题。
[4]反应条件温和,避免了高温高压以及超低温的反应条件,非常适合工业化方法处理;[5]整个工艺中大部分溶剂都可以回收,三废产生量少,可以视为清洁生产工艺;[6]总之,采用本工艺的生产工艺,经济效益和社会效益都十分的显著。
四、具体实施例[步骤1] 实施例1[在2L的三口瓶中加入腺嘌呤100g (0. 74mo1),充N2保护,然后加入500m1的DMF,开启搅拌,加入,加入R一碳酸丙烯酯95g (0. 92mo1),加入NaOH 3g (0. 075mo1),室温搅拌10分钟,体系均匀后,开始升温至100一130℃,保温反应,12小时以后取样检测,(反应清澈透明);当腺嘌呤在反应体系中含量在1%以下,可以停止反应;慢慢降至90,C以下,开始加入甲苯600m1,有大量白色固体析出;降温至室温后,继续降至0-5℃,保温搅拌2小时;抽滤,用200m1冰甲苯泡洗滤饼,得到湿滤饼约180g,干燥后得品R-9- (2-羟基丙基)腺嘌呤约130-140g,HPLC纯度85% -90%,熔点188一192℃;精制:将上步得到的R-9- (2-羟基丙基)腺嘌呤产物130g加入600m1的乙醇,100m1的甲苯,升温至溶清,加入2g的活性炭,回流半小时后趁热抽滤除去活性炭,滤液降至室温,进一步冷却至0℃,保温1小时后抽滤,干燥后得到105g左右的产物R-9- (2-羟基丙基)腺嘌呤,HPLC纯度98%以上,mol收率73%左右。
[步骤1]实施例2[0070]在2L的三口瓶中加入腺嘌呤100g (0. 74mo),充N2保护,然后加入700m1的DMSO,开启搅拌,加入,加入R一碳酸丙烯酯95g (0. 92mo1),加入KOH 4g (0. 071mo1),室温搅拌10分钟,体系均匀后,开始升温至110一125℃,保温反应,8小时以后取样检测,反应基本清澈或带少许絮状物;当腺嘌呤在反应体系中含量在1%以下,可以停止反应;慢慢降温至室温后,继续降至0-5℃,保温搅拌2小时;抽滤,用200m1冰甲苯泡洗滤饼,得到湿滤饼约170g,干R-9- (2-羟基丙基)腺嘌呤约125-130g,HPLC纯度82%-88%,熔点182-1880C;[00711精制:将上步得到的产物,130g加入500m1的甲醇,100m1的甲苯,升温至溶清,加入5g的活性炭,回流半小时后趁热抽滤除去活性炭,滤液降至室温,进一步冷却至0℃,保温1小时后抽滤,干燥后得到101g左右的产物R-9- (2-羟基丙基)腺嘌呤,HPLC纯度98. 5%以上,mol收率70%左右。
[步骤2]实施例3[在1L干燥过的四口烧瓶中,加入R-9- (2-羟基丙基)腺嘌呤100g (0. 52mol),充N2保护,加入DMF200m1,开启搅拌;加入叔丁醇镁90g(0. 53mo1),整个反应体系呈灰白色浑浊,(叔丁叔丁醇镁遇水分解,需保证整个体系无水无氧);慢慢升温至50一70℃,在此温度下保温1小时;然后升温至60一80℃,开始滴加250gDESMP(对甲苯磺酰氧基磷酸二乙酯)(0. 77mo l );随着DESMP的滴加,反应液会放热,控制滴加速率,约1.5hrs滴完,当DESMP 滴加完75%左右时,反应液会变成清澈胶状透明橙黄色;滴加完2小时后检测HPLC,当HPLC分析原料中R-9- (2-羟基丙基)腺嘌呤在2%以下即可停止反应;慢慢降至室温,滴加75g(1.24mol)无水乙酸,约15min左右滴加完毕;滴加完毕后,室温搅拌30分钟;将反应液转移至另一2L的三口烧瓶中,磁力搅拌,油浴减压蒸馏;用减压蒸出DMF,最高内温控制在100℃以下,2小时左右蒸干;蒸除完毕后,将油浴降至40℃左右,保证瓶内物料不固化;加入1000m1二氯甲烷,自然冷却至25-30℃,换机械搅拌30分钟;加入100m1的水,有大量白色固体析出,保温搅拌1小时;抽滤过滤去固体,得到固体240g(湿品,应为丙酸镁),(可用二氯甲烷洗涤);静置分二层后,将二氯甲烷层浓缩至干,得到第二步产物R-9- [2-(二乙基磷酰甲氧基)丙基〕腺嘌呤180g,为淡黄色油状物,纯度98%以上,mol 收率95%左右。