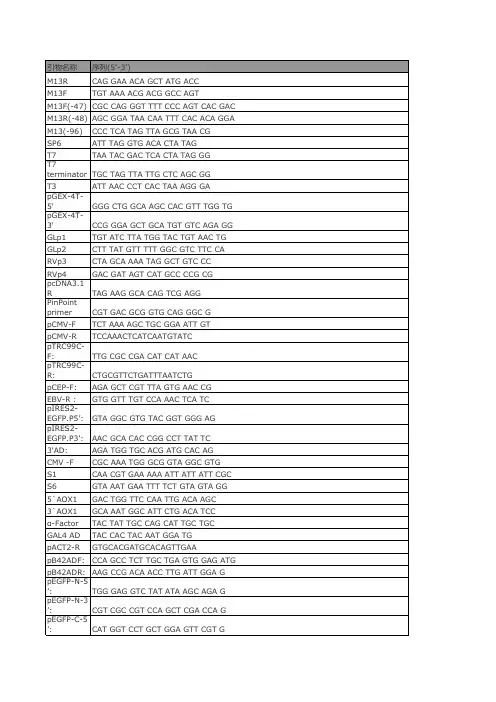
简并引物列表
- 格式:pdf
- 大小:83.93 KB
- 文档页数:1



第27卷第4期2008年7月 食品与生物技术学报Journal of Food Science and Biotechnology Vol.27 No.4J ul. 2008 文章编号:167321689(2008)0420091206 收稿日期:2007210225. 基金项目:国家自然科学基金项目(30570142).作者简介:陈献忠(19802),男,安徽界首人,工业微生物博士研究生.3通讯作者:诸葛健(19392),男,浙江金华人,教授,博士生导师,主要从事发酵工程和工业微生物学方面的研究.Email :jianzhuge @优化简并引物PCR 克隆Can di da gl yceri nogenes32磷酸甘油脱氢酶基因片段陈献忠, 饶志明, 沈微, 诸葛斌, 方慧英, 王正祥, 诸葛健3(江南大学工业生物技术教育部重点实验室,江苏无锡214122)摘 要:为了从工业生产菌株耐高渗产甘油假丝酵母(Can di da gl yceri nogenes )克隆甘油合成的限速酶编码基因胞浆NAD +232磷酸甘油脱氢酶基因(ct GPD ),对不同酵母和其他真核生物的NAD +232磷酸甘油脱氢酶进行比对,分析氨基酸和核苷酸的保守序列,设计了4对简并引物用于扩增C.gl yceri nogenes 的NAD +232磷酸甘油脱氢酶(GPD )基因片段,经过优化PCR 反应条件,利用其中一对中等简并度的引物扩增出Cg GPD 基因中约600bp 的保守核心片段。
DNA 序列及推绎的氨基酸序列进行比对分析表明,该基因片段与其他酵母的胞浆NAD +232磷酸甘油脱氢酶基因的对应区域具有典型的保守区域,并且与安格斯毕赤酵母的GPD 基因相似性较高。
关键词:PCR ;NAD +依赖32磷酸甘油脱氢酶;产甘油假丝酵母;克隆;简并引物中图分类号:Q 785文献标识码:ACloning of NAD +2Dependent G lycerol 32Phosphate DehydrogenaseG ene Fragment from Ca ndida gl yceri nogenes withDegenerate Oligonucleoide PrimersC H EN Xian 2Zhong , RAO Zhi 2Ming , SH EN Wei , ZHU GE Bin ,FAN G Hui 2Y ing , WAN G Zheng 2Xiang , ZHU GE Jian 3(Key Lab of Industrial Biotechnology ,Ministry of Education ,Jiangnan University ,Wuxi 214122,China )Abstract :In S accharom yces cerevisi ae ,glycerol is synt hesized in cytosol by reduction of t he glycolytic intermediate dihydroxyacetone p ho sp hate (D HA P )to glycerol 32p hosp hate (G3P )followed by dep hosp horylation of G3P to glycerol.Furt her st udies had revealed t hat cytol NAD +2dependent glycerol 32p ho sp hate dehydrogenase (ct GPD )is t he rate 2limiting enzymes for glycerol synt hesis.However ,it is unclear whet herGPD has similar role in Candi dagl yceri nogenes WL200225,a novel o smotolerant yeast species used in glycerol p roduction.So ,itis essential to clone GPD gene from C.gl yceri nogenes ,for investigation of glycerol bio synt hesis in molecular level.After comparison of t he amino acid sequence of t he GPD H p roteins in several species ,degenerate oligonucleoide p rimers were designed for PCR based on conservative regions in t he amino acid sequence.PCR was performed on t he genomic DNA of C.gl yceri nogenes andparameters involved in amplification efficiency were optimized.U sing one moderate degeneracy pair of p rimers,a degenerate PCR p roduct of ca.0.6kb was amplified and sequenced.Which homologous to GPD1of S accharom yces cerevisi ae.This provides a t hreshold of st udying glycerol p roduction pat hway and osmotic st ress response mechanism at genetic and molecular level in t his yeast.K ey w ords:PCR;NAD+2dependent glycerol32p ho sp hate dehydrogeanse;Candi da gl yceri nogenes;clone;degenerate oligonucleoide primers 当细胞处于高渗透压环境中时,为了保持胞内的水分和小分子物质向环境中外泄,就需要通过自身合成一些生物相容性物质来提高胞内的渗透压来平衡胞内和胞外的压力差,从而起到保护和防御的作用。


常用引物序列引言引物序列是指在分子生物学实验中用来特异性引导DNA或RNA复制和扩增的短链核酸序列。
常用的引物序列在科研和实验室工作中起着重要作用。
本文将介绍几种常用引物序列及其在实验中的应用。
1. 通用引物序列通用引物序列是指适用于多种物种的引物序列。
常见的通用引物序列包括16S rRNA引物序列用于细菌和古细菌的分析,18S rRNA引物序列用于真核生物的分析,以及ITS引物序列用于真菌的分析。
这些通用引物序列具有高度保守性,可以用于不同物种的物种鉴定和进化分析。
2. 物种特异引物序列物种特异引物序列是指只在特定物种中存在的引物序列。
这些引物序列通常用于物种鉴定和种群遗传分析。
例如,人类特异引物序列可以用于人类DNA的特异扩增,从而进行人类个体的鉴定。
物种特异引物序列的选择和设计需要根据目标物种的基因组序列进行。
3. 定量引物序列定量引物序列是指用于定量PCR实验的引物序列。
这些引物序列通常与目标基因的外显子区域相结合,可以通过PCR扩增目标基因的特定片段,并通过实时荧光定量PCR技术进行定量分析。
定量引物序列的设计需要考虑引物的特异性和扩增效率,以确保准确的定量结果。
4. 荧光标记引物序列荧光标记引物序列是指在引物序列上加入荧光标记分子,用于检测和定量特定基因的表达水平。
常用的荧光标记引物序列包括荧光探针和荧光引物。
荧光标记引物序列可以通过荧光定量PCR或原位杂交等技术进行检测。
5. 反向引物序列反向引物序列是指用于反转录PCR实验的引物序列。
反转录PCR是一种将RNA转录成cDNA的技术,常用于研究基因的表达调控和mRNA的定量分析。
反向引物序列通常与反向转录酶结合,将RNA逆转录成cDNA,并通过PCR扩增特定基因的cDNA片段。
6. 基因特异引物序列基因特异引物序列是指用于特定基因扩增的引物序列。
这些引物序列通常与目标基因的外显子区域相结合,可以选择性地扩增目标基因,从而进行基因型鉴定和基因表达分析。
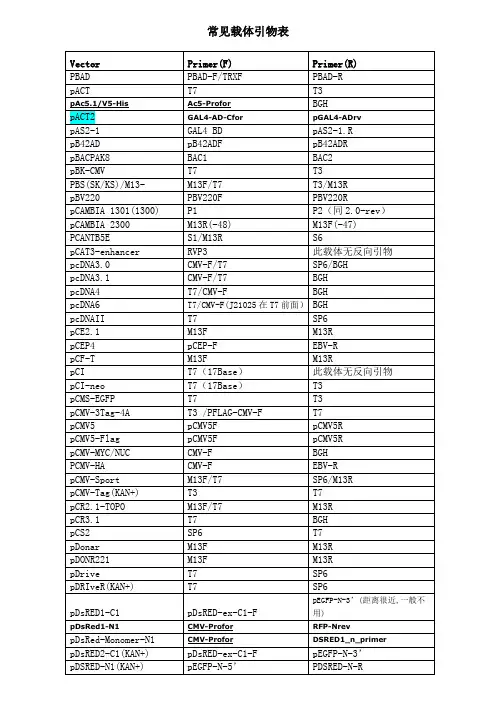


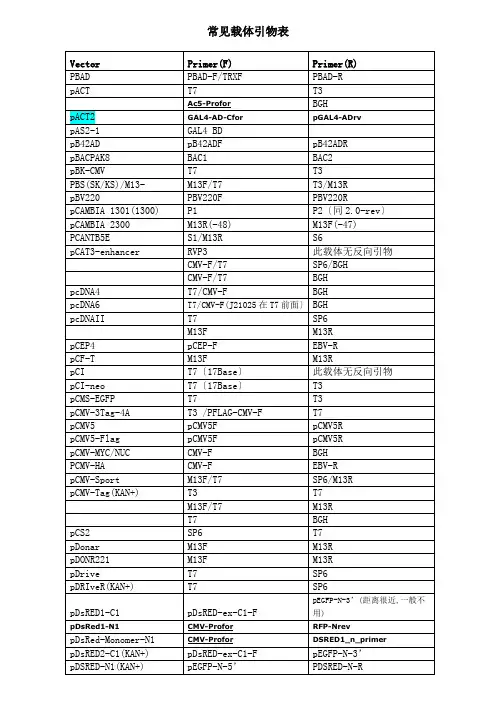
常用引物序列(分子生物学)序列(5'-3') GGTTACCTTGTTACGACTT AGAGTTTGATCCTGGCTCA GACGCACAATCCCACTATCC AGATGGTGCACGATGCACAG GGCAAATGGCATTCTGACAT TAAGAGTCACTTTAAAATTTGTATAC TACCACTACAATGGATGATG GACTGGTTCCAATTGACAAGC TCATCGGAAGAGAGTAG CCCTCATAGTTAGCGTAACG TACTATTGCCAGCATTGCTGC AACCATCTCGCAAATAAATA ACGCACAGAATCTAGCGCTT TAGAAGGCACAGTCGAGG TTAGGACAAGGCTGGTGG ATAACCCCGCCCCGTTGCGCAAATGGGCGGTAGGCGTG\ATGGGCGGTAGGCGT G TCGTTGGGCGGTCAGC GATTATGCGGCCGTGTACAA GATCTCGACGCTCTCCCT TTGTACACGGCCGCATAATC GTGGTTTGTCCAAACTCATC AGCACCCAGTCCGCCCTGAGC CGTCGCCGTCCAGCTC AACATTTTCGGTTTGTATTACTTC TGTATCTTATGGTACTGTAACTG CTTTATGTTTTTGGCGTCTTCCA GLP4 H1-F\H1 HSVtk-pArev IRES-R ITS4 ITS5 M13-47M13F\TrxReverse\M13-20\M13F-4 0 M13R malE MT-Profor mU6F2(mouse) NL1 NL4 NOSR-primer P1 p10-Profor P2 pBABE3' PBABE5' pBADfw pBADrv\pTrcHis-rv PBV220F pCDNA3.1F pCMV5F pCMV5R PCMV-F PCMV-R GCCCTTCTTAATGTTTTTG TCGCTATGTGTTCTGGGAAAGTCTCCTTCCGTGTTTCAG CCTCACATTGCCAAAAGACG TCCTCCGCTTATTGATATGC GGAAGTAAAAGTCGTAACAAGG CGCCAGGGTTTTCCCAGTCACGAC TGTAAAACGACGGCCAGT CAGGAAACAGCTATGACC GGTCGTCAGACTGTCGATGAAGCC CATCTCAGTGCAACTAAA TAGTATCCGACGCCGCCATC GCATATCAATAAGCGGAGGAAAAG GGTCCGTGTTTCAAGACGG CGTCCAGCTCCATGTTGC CCAGGCTTTACACTTTATGC CGGACCTTTAATTCAACCC GCGATTAAGTTGGGTAACGC ACCCTAACTGACACACATTCC CTTTATCCAGCCCTCAC CCATAGCATTTTTATCCATAAG ATCTGTATCAGGCTGAAAATC AAGAAGGGCAGCATTCAAAG CTAGAGAACCCACTGCTTAC TTCCAAAATGTCGTAATAAC ATTATAGAGGACACCTAGTC TCTAAAAGCTGCGGAATTGT TCCAAACTCATCAATGTATCpcoldI-R\pCold-SUMO-R pCold-TF-F1 pDBLeu-F pDEST22-F pDEST22-R PDONR-F PDONR-R pDsRED-ex-C1-FPEF-F\EF-1aForward pEGFP-C-3' pEGFP-C-5’ pEGFP-N-3’\EGFP-Nrev pEGFP-N-5'pFastBac-fw pFastBac-R pFastBac-rev pFlag-CMV-F pFlag-CMV-R PGEX-F PGEX-R PH-Profor pIRESrv PJET1.2-F PJET1.2-R pLNCX-F pLXSN-F pLXSN-R pMAL-C2X-FpMAL-C2X-RGGCAGGGATCTTAGATTCTG CCACTTTCAACGAGCTGATGGAATAAGTGCGACATCATCATC TCGATGATGAAGATACCCCACC CTCGACGTCTTACTTACTTAGC TCGCGTTAACGCTAGCATGGATCTC GTAACATCAGAGATTTTGAGACAC TCCCACAACGAGGACTACAC TCAAGCCTCAGACAGTGGTTC TATGGCTGATTATGATCAGT CATGGTCCTGCTGGAGTTCGTG CGTCGCCGTCCAGCTCGACCAG TGGGAGGTCTATATAAGCAGAG GTCCGAAACCATGTCGTAC TGGACAAACCACAACTAGAATG CCTCTACAAATGTGGTATGG GGTAGGCGTGTACGGTGG GCACTGGAGTGGCAACTT GGCAAGCCACGTTTGGTG GAGCTGCATGTGTCAGAGG AAATGATAACCATCTCGC GCCCTAGATGCATGCTCG CGACTCACTATAGGGAGAGCGGC AAGAACATCGATTTTCCATGGCAG AGCTCGTTTAGTGAACCGTCAGATCG CTTGAACCTCCTCGTTCGAC GTTGCTGACTAATTGAGATG TGCGTACTGCGGTGATCAAC CTGCAAGGCGATTAAGTTGGpMAL-C5x-R PPC86-F\pDEST22-F2018 PPC86-R\pDBLeu-R pQE30-F pQE30-R pSHUTTLE-CMV-R pSilencer4.1-F\2.0rev PTRC99C-F PTRC99C-R\PBV220R pYes2.R\CYC1-Terminator RFP-Nrev RV-M RVP3 RVP4 S.tag SeqL-A SeqL-B SP6 SV40 SV40-pArev\pSG5rv T3 T7 T7(17base) T7terU6-promoter(human) v1.5 V5rev WPRE-R XL39\CMV-24 TGTCCTACTCAGGAGAGCGTTCAC TATAACGCGTTTGGAATCACT GTAAATTTCTGGCAAGGTAGAC TGAGCGGATAACAATTTCACGTTCTGAGGTCATTACTGG GTGGTATGGCTGATTATGATCAG AGGCGATTAAGTTGGGTA TTGCGCCGACATCATAAC CTGCGTTCTGATTTAATCTG GCGTGAATGTAAGCGTGAC GTTCACGGTGCCCTCC AGCGGATAACAATTTCACACAGGA CTAGCAAAATAGGCTGTCCC GACGATAGTCATGCCCCGCG GAACGCCAGCACATGGAC GCAGTTCCCTACTCTCGC CATCAGAGATTTTGAGACAC ATTTAGGTGACACTATAG GGAGGCTTTTTTGGAGGC GAAATTTGTGATGCTATTGC ATTAACCCTCACTAAAGGGA TAATACGACTCACTATAGG ACATCCACTTTGCCTTTCTC TGCTAGTTATTGCTCAGCGG CCGTAACTTGAAAGTATTTCG GGACTTTCCAAAATGTCG ATCGAGACCGAGGAGAGG CATAGCGTAAAAGGAGCAACA ATTAGGACAAGGCTGGTGGGITS1TCCGTAGGTGAACCTGCGG。

简并引物设计PCR(聚合酶链反应)是一种基础而重要的技术,在生物学、医学、检测等领域都有着广泛的应用。
然而,PCR技术在实际应用过程中,常常会遇到一些问题,例如引物设计不合理、多重特异性等。
简并引物设计便是为了解决这些问题而产生的技术。
本文将介绍简并引物设计的原理、方法、优缺点及应用。
一、简并引物设计的原理简并引物是一种含有多个碱基的引物,其中每个碱基的位置都可以存在多种可能性。
简并引物的设计原理是根据目标序列的多态性,将可能出现的碱基变异情况考虑在内,从而设计出能够特异性扩增目标序列的引物。
二、简并引物设计的方法简并引物设计可以通过人工设计或计算机辅助设计完成。
1. 人工设计人工设计简并引物需要根据目标序列的多态性情况,逐个考虑每个碱基的变异情况,然后选择合适的碱基组合作为简并引物的设计。
例如,对于一段序列“ATGCTAGC”,在第三个碱基位置上存在两种可能性“A”和“T”,在第七个碱基位置上存在三种可能性“A”、“C”和“G”,则可以设计出两个简并引物:ATGCTAGC和ATGCTAGN。
2. 计算机辅助设计计算机辅助设计可以通过在线工具或软件完成,例如Primer3、Primer-BLAST等。
这些工具可以根据用户输入的目标序列信息和扩增条件,自动生成合适的简并引物设计方案。
三、简并引物设计的优缺点简并引物设计相比于传统引物设计有以下优点:1. 提高特异性简并引物设计可以考虑到目标序列的多态性,从而提高扩增的特异性。
特别是对于高度变异的序列,简并引物设计可以有效地避免非特异性扩增。
2. 减少设计时间传统引物设计需要逐个考虑每个碱基的变异情况,而简并引物设计可以通过在线工具或软件自动生成设计方案,大大减少了设计时间。
3. 降低成本简并引物设计可以减少试验重复次数和材料浪费,从而降低实验成本。
简并引物设计的缺点包括:1. 扩增效率可能降低由于简并引物设计需要考虑目标序列的多态性,因此引物长度可能会增加,扩增效率可能会降低。
简并引物引言简并引物是一种在分子生物学研究和实验室技术中常用的工具。
简并引物是一组具有部分序列差异但具有相似特性的引物序列,用于扩增目标DNA序列。
简并引物通常由多个互补碱基序列组成,这些引物序列可以与目标DNA序列的多个可能序列匹配。
简并引物的广泛应用促进了分子生物学研究的发展,并在基因组学、遗传学和医学诊断中扮演着重要的角色。
简并引物的设计原理简并引物的设计原理基于核酸序列间的碱基互补配对。
在DNA复制和PCR扩增的过程中,引物需要与DNA模板中的特定序列互补,使得DNA聚合酶能够以引物为起始点开始合成新的DNA链。
为了确保引物能够选择性地与目标DNA序列互补,并且不与其他非目标序列互补,简并引物的设计需要考虑到以下几个因素:1.碱基互补:引物序列中的碱基需要与目标DNA序列完全互补,以确保引物能够牢固地结合并启动DNA链的合成过程。
2.简并性:引物序列需要包含多个互补碱基序列,以使得引物能够与目标DNA序列的多个可能序列匹配。
这样的设计可以增加引物与目标DNA序列杂交的特异性和选择性。
3.长度:引物的长度需要适当,一般为15-25个碱基。
过短的引物可能导致非特异性引物杂交,而过长的引物可能影响反应的效率。
4.碱基组成:引物的碱基组成需要均衡,避免引物序列中存在偏向某种碱基的情况。
碱基组成的不均衡可能导致引物的特异性降低。
任何一个简并引物的设计都需要综合考虑以上因素,以确保引物能够准确、特异地扩增目标DNA序列。
简并引物的应用简并引物具有广泛的应用领域,包括但不限于以下几个方面:1.PCR扩增:PCR是分子生物学中最常用的实验技术之一,简并引物在PCR扩增中起到至关重要的作用。
通过使用简并引物,可以扩增出多个不同但相关的目标DNA片段,从而快速、准确地分析目标序列的多态性和变异情况。
2.遗传标记:简并引物可以用于遗传标记的设计和分析。
通过引物的简并性,可以覆盖目标DNA序列的各种可能序列,从而提高遗传标记的准确性和信息量。
HPV病毒的常用通用引物检测摘要研究表明,HPV病毒感染与人体多种肿瘤的发生相关。
对感染个体的病毒检测具有重要的疾病诊断、治疗、预防意义。
但目前缺乏可以兼顾敏感性、特异性、操作简单,可以大规模推广使用的可靠检测方法。
对待测样本进行PCR 检测仍是目前实验室最多采用的检测方法。
PCR具有高敏感、易操作的优点,但其反应的灵活性和极高的敏感性,也带来了假阳性和假阴性的问题。
针对HPV 病毒L1序列保守区域设计的通用引物检测,最常被应用于大样本检测。
通用引物可以在一次PCR反应中同时检测多种型别的HPV病毒感染,与序列特异PCR 检测相比,可以大大减少检测的工作量。
但其缺点在于,通用引物仅与某个或少数HPV型序列匹配,而对于多数型别HPV,引物结合区都存在或多或少的碱基错配。
这使得通用引物对不同型别的HPV扩增效率不同。
因此,使用通用引物检测HPV感染时,对于某些与引物匹配较差的HPV型别,其感染情况往往可能被低估。
目前常用的通用引物根据设计原则不同分为三类:1)单一引物如:GP5+/6+。
2)简并引物如:、MY09/11。
3)混合引物如:SPF1/2。
个型通用引物都有其各自的优缺点,需要根据特定试验的目的、条件选用合适的通用引物进行检测。
【关键词】HPV病毒;通用引物;单一引物;简并引物;混合引物HPV病毒是一种亲上皮细胞的双链DNA病毒,其基因组长约7900bp。
目前约共发现约200多型HPV病毒。
该病毒主要感染皮肤和粘膜的上皮细胞,造成上皮增生甚至恶性转化[1] [2] [3]。
人们已经在生殖道检测到超过40型的HPV,其中许多与宫颈内皮增生及宫颈癌密切相关,如:16、18、31、33、35等,这部分HPV病毒被称为高危型[4][5]。
而HPV6、11等型别主要存在于良性病变中,多造成生殖道疣等疾病,被称为低危型[6][7]。
但人为区分的高危型和低危型并不是绝对的,因为在一些癌变中确实也存在所谓的低危型HPV[8]。
鸟类1.雀形目38种鸟类线粒体cytb引物:通过软件设计, 得到了cytb 的通用引物, 引物序列为: 5‘- CCTACTTAGGATCATTCGCCCT – 3’和5‘-GTCTTCATCTCCGGTTTACAAGAC – 3‘。
7.鹤的性别鉴定8.鸟类的通用引物(我国部分家鸭品种的DNA条形码初步分析)两栖类通用引物龟类3.亚洲淡水和陆生龟鳖类12S r R N A 基因片段的序列分析和系统发生研究PC R 扩增所用引物为L 1 0 9 1 (5 ’一A A A A A G C T T C A A A C T G G G A T T A G A T A C C CC A C T A T 一3 ’)和H 1 4 7 8 (5 ’一T G A C T G C A G A G G G T G A CG G G CG G T G T G T 一3 ’) (K o e he r e t a l. , 1 9 8 9 ) ,L、H 分别为轻链和重链, 数字与人m tD N A 序列对应P C R 反应体积为1 0 0 拜l, 反应液中含1 om m o l/ L T r is 一H C I, p H 5 . 3 , 5 0 m m o l/ L K C I, 0. 1 % T r ito nx一1 0 0 , 2 .5 m m o l/ L Mg C 12 , 2 0 0 拜m o l/ L 4 种d N T P , 1 0 p m o l z 条引物, zU T a q 酶。
p C R 反应在PE 2 4 0 型PC R 仪上进行, 循环参数为95 ℃变性40 秒, 5 ℃退火60 秒, 72 ℃延伸60 秒, 循环次数为3 5 次。
4.福尔马林保存的龟鳖类动物基因组DNA的提取方法5.Limited efficiency of universal mini-barcode primers for DNA amplification from desert reptiles, birds and mammalsThe mini-barcode (130 bp) was amplified using the primer pair Uni-MinibarF1 (TCC ACT AAT CAC AAR GAT ATT GGT AC) and Uni-MinibarR1 (GAA AAT CAT AAT GAA GGC ATG AGC), as reported earlier (Meusnier et al., 2008). The reactants contained 12 mL FideliTaq PCR master mix (USB Corporation, USA), template DNA (2 mL), each primer (50 nmol in 0.5 mL) in a reaction volume of 25 mL. Besides the standard protocol based on two sequential annealing temperatures (Meusnier et al 2008), we also tested the PCR amplifications using a more stringent (single-annealing) protocol. The thermal cycling conditions for both these protocols are summarized in Table 2. The PCR products were electrophoresed on a 1% agarose gel stained with ethidium bromide. The bands in the gel were visualized under UV light and evaluated using a Proxima C16 Phi+ gel imaging system (Isogen Life Science, The Netherlands昆虫本研究运用常规的DNA条形码通用引物(Lep-F1, 5’-ATTCAACCAATCATAAAGA TA T-3’; 和Lep-R1, 5’-TAAACTTCTGGA TGTCCAAAAA-3’) (Hebert et al., 2004a),得到片断长度为652bp。
混合简并引物1. 简介混合简并引物是一种在分子生物学研究中广泛应用的技术,用于扩增特定DNA序列。
通过使用多个引物的混合,可以提高扩增反应的特异性和灵敏度,从而增加目标序列的检测准确性和成功率。
本文将介绍混合简并引物的原理、设计方法和应用领域。
2. 原理混合简并引物的设计基于引物的碱基序列和碱基对的配对规则。
在DNA扩增反应中,引物与目标DNA序列的两个互补部分结合,形成一个稳定的双链结构。
引物的碱基序列决定了扩增反应的特异性和选择性。
通过引入简并位点,即允许多个碱基在同一位点出现,可以增加引物的变异性,从而提高目标序列的扩增准确性。
混合简并引物的设计过程包括以下步骤:1.确定目标DNA序列:根据研究需要,确定待扩增的目标DNA序列。
2.引物设计:根据目标DNA序列,设计一组简并引物,其中包含多个简并位点。
简并位点可以使用IUPAC碱基代码表示,例如R表示A或G,S表示C或G。
3.引物评估:使用计算工具或软件评估引物的特异性和选择性。
确保引物与目标DNA序列的互补部分匹配,并避免与非目标序列的互补部分匹配。
4.引物合成:将设计好的引物合成,并进行质量检测。
3. 设计方法混合简并引物的设计方法可以根据目标DNA序列的长度和复杂性进行调整。
以下是常用的设计方法:1.单简并位点设计:适用于目标DNA序列较短且没有复杂结构的情况。
在引物的特定位点引入一个简并位点,例如使用Y表示C或T。
这种方法简单快捷,但对于复杂的目标序列可能不够灵活。
2.多简并位点设计:适用于目标DNA序列较长或具有复杂结构的情况。
在引物的多个位点引入简并位点,可以使用多个不同的IUPAC码。
例如,可以使用R、Y、S等表示不同的碱基组合。
这种方法可以增加引物的变异性,提高扩增反应的特异性。
3.引物长度调整:根据目标DNA序列的长度和复杂性,调整引物的长度。
较长的引物可以提高特异性,但也增加了非特异性扩增的风险。
4. 应用领域混合简并引物在分子生物学研究中有广泛的应用,包括:1.基因突变检测:通过扩增目标基因的特定区域,可以检测和鉴定基因突变。
HPV病毒的常用通用引物检测摘要研究表明,HPV病毒感染与人体多种肿瘤的发生相关。
对感染个体的病毒检测具有重要的疾病诊断、治疗、预防意义。
但目前缺乏可以兼顾敏感性、特异性、操作简单,可以大规模推广使用的可靠检测方法。
对待测样本进行PCR 检测仍是目前实验室最多采用的检测方法。
PCR具有高敏感、易操作的优点,但其反应的灵活性和极高的敏感性,也带来了假阳性和假阴性的问题。
针对HPV 病毒L1序列保守区域设计的通用引物检测,最常被应用于大样本检测。
通用引物可以在一次PCR反应中同时检测多种型别的HPV病毒感染,与序列特异PCR 检测相比,可以大大减少检测的工作量。
但其缺点在于,通用引物仅与某个或少数HPV型序列匹配,而对于多数型别HPV,引物结合区都存在或多或少的碱基错配。
这使得通用引物对不同型别的HPV扩增效率不同。
因此,使用通用引物检测HPV感染时,对于某些与引物匹配较差的HPV型别,其感染情况往往可能被低估。
目前常用的通用引物根据设计原则不同分为三类:1)单一引物如:GP5+/6+。
2)简并引物如:、MY09/11。
3)混合引物如:SPF1/2。
个型通用引物都有其各自的优缺点,需要根据特定试验的目的、条件选用合适的通用引物进行检测。
【关键词】 HPV病毒;通用引物;单一引物;简并引物;混合引物HPV病毒是一种亲上皮细胞的双链DNA病毒,其基因组长约7900bp。
目前约共发现约200多型HPV病毒。
该病毒主要感染皮肤和粘膜的上皮细胞,造成上皮增生甚至恶性转化[1] [2] [3]。
人们已经在生殖道检测到超过40型的HPV,其中许多与宫颈内皮增生及宫颈癌密切相关,如:16、18、31、33、35等,这部分HPV 病毒被称为高危型[4][5]。
而HPV6、11等型别主要存在于良性病变中,多造成生殖道疣等疾病,被称为低危型[6][7]。
但人为区分的高危型和低危型并不是绝对的,因为在一些癌变中确实也存在所谓的低危型HPV[8]。