报错解决
- 格式:doc
- 大小:437.94 KB
- 文档页数:13

文章标题:深入探讨C++编程中报错error的解决方法在C++编程中,我们经常会遇到各种各样的报错信息,这不仅是初学者的困扰,即便是经验丰富的程序员也会遇到各种报错情况。
在本文中,我将为您详细解读C++编程中常见的报错类型,并提供解决方法,帮助您更好地理解和应对这些问题。
1. 编译错误编译错误是在编译期间出现的错误,通常指程序无法通过编译器的检查,无法生成可执行文件。
常见的编译错误包括语法错误、语义错误和类型错误。
我们可以通过查看编译器的报错信息,逐行检查代码,找出并修复错误所在。
2. 运行时错误运行时错误是指程序在运行过程中出现的错误,导致程序异常终止或产生错误结果。
常见的运行时错误包括空指针引用、数组越界访问、未捕获的异常等。
我们应该在编写代码时加入适当的防御性编程,对可能出现的异常情况进行处理和容错。
3. 逻辑错误逻辑错误是指程序在逻辑上有误,导致程序无法按照预期的逻辑执行。
通常这类错误不会导致程序崩溃,但会导致程序输出错误的结果。
在遇到逻辑错误时,我们可以通过调试工具逐步执行程序,定位错误所在,并修复逻辑错误。
4. 解决方法针对不同类型的报错,我们可以采取相应的解决方法。
在面对编译错误时,我们需要仔细查看编译器的报错信息,逐行检查代码,修复语法、语义和类型错误;对于运行时错误,我们需要在代码中加入适当的异常处理和错误检测机制,确保程序能够处理各种异常情况;而在面对逻辑错误时,则需要通过调试工具逐步执行程序,找出逻辑错误的根源并加以修复。
总结与回顾通过本文的内容,我希望您能更清晰地理解C++编程中常见报错的类型和解决方法。
无论是编译错误、运行时错误还是逻辑错误,都不是令人畏惧的难题,只要我们用心对待,认真分析和解决问题,就能够在编程的道路上走得更远。
个人观点与理解在我看来,C++编程中遇到报错并非坏事,反而可以帮助我们发现程序中潜在的问题,提高代码的质量和健壮性。
通过及时解决报错问题,我们能够更好地理解代码运行的机制,提升自己的编程水平。

3dsll报错的解决方法3dsll报错是指任天堂3DS LL主机在使用过程中出现错误提示或无法正常运行的问题。
这些错误可能涉及软件、硬件、网络等多个方面。
下面将列举一些常见的3dsll报错并提供解决方法。
一、软件错误相关报错及解决方法:1. 系统错误:可能是由于系统文件损坏、升级失败等原因导致。
解决方法如下:a. 尝试重启主机,看是否可以解决问题。
b. 如果无法解决,则可以考虑重置主机的系统设置。
c. 如果问题仍然存在,可以尝试联系任天堂客服寻求帮助或寄修。
2. 游戏卡无法读取:这种情况下主机可能无法正确读取游戏卡带。
解决方法如下:a. 检查游戏卡是否有损坏或脏污。
如果有,可以尝试用软布擦拭游戏卡金属接触点,并再次尝试读取。
b. 如果问题仍然存在,可以重新插拔游戏卡,确保插入正确并牢固。
3. 无法下载游戏或更新:这种情况下可能是网络连接问题或系统设置问题。
解决方法如下:a. 检查网络连接是否正常,尝试重新连接Wi-Fi。
b. 检查主机Wi-Fi设置是否正确,尝试重新设置。
c. 如果问题仍然存在,可以尝试重启主机再次下载游戏或更新。
4. 屏幕闪烁或黑屏:这种情况下可能是主机显示屏幕出现问题导致。
解决方法如下:a. 尝试将主机关机,然后插拔电池重新启动。
b. 检查主机是否已经电量不足,如果是则充电后再次尝试开机。
c. 如果问题仍然存在,可以尝试联系任天堂客服寻求帮助或寄修。
二、硬件错误相关报错及解决方法:1. 触摸屏失灵:这种情况下可能是主机触摸屏出现问题导致。
解决方法如下:a. 尝试关机,然后用软布清洁触摸屏,并再次尝试使用。
b. 检查主机是否存在硬件损坏,如触摸屏线路损坏。
如果有,则需要联系任天堂客服寻求帮助或寄修。
2. 按键失灵或卡键:这种情况下可能是主机按键出现问题导致。
解决方法如下:a. 尝试关机,然后用软布清洁按键,检查是否有物体进入按键造成卡键。
再次尝试使用。
b. 检查主机是否存在硬件损坏,如按键弹性变差或损坏。

10种常见B IOS报错故障解决方法开机自检时出现问题后会出现各种各样的英文短句,短句中包含了非常重要的信息,读懂这些信息可以自己解决一些小问题,可是这些英文难倒了一部分朋友,下面是一些常见的BIO S短句的解释,大家可以参考一下。
1.CMOS batter y failed中文:CMOS电池失效。
解释:这说明CMO S电池已经快没电了,只要更换新的电池即可。
2.CMOS checksum error-Defaul ts loaded中文:CMOS执行全部检查时发现错误,要载入系统预设值。
解释:一般来说出现这句话都是说电池快没电了,可以先换个电池试试,如果问题还是没有解决,那么说明CM OSRAM可能有问题,如果没过一年就到经销商处换一块主板,过了一年就让经销商送回生产厂家修一下吧!3.PressESC to skip memory test中文:正在进行内存检查,可按ESC键跳过。
解释:这是因为在C MOS内没有设定跳过存储器的第二、三、四次测试,开机就会执行四次内存测试,当然你也可以按ESC键结束内存检查,不过每次都要这样太麻烦了,你可以进入C OMS设置后选择BI OSFEA TURSS ETUP,将其中的Qu ick PowerOn Self Test设为Enabl ed,储存后重新启动即可。
4.Keyboa rd erroror no keyboa rd presen t中文:键盘错误或者未接键盘。
解释:检查一下键盘的连线是否松动或者损坏。
5.Hard disk instal l failur e中文:硬盘安装失败。
解释:这是因为硬盘的电源线或数据线可能未接好或者硬盘跳线设置不当。
你可以检查一下硬盘的各根连线是否插好,看看同一根数据线上的两个硬盘的跳线的设置是否一样,如果一样,只要将两个硬盘的跳线设置的不一样即可(一个设为Ma ster,另一个设为S lave)。

报错解决V ASP⾃旋轨道耦合计算错误汇总静态计算时,报错:VERY BAD NEWS! Internal内部error in subroutine⼦程序IBZKPT:Reciprocal倒数的lattice and k-lattice belong to different class of lattices. Often results are still useful (48)INCAR参数设置:对策:根据所⽤集群,修改INCAR中NPAR。
将NPAR=4变成NPAR=1,已解决!错误:sub space matrix类错误报错:静态和能带计算中出现警告:W ARNING: Sub-Space-Matrix is not hermitian共轭in DA V结构优化出现错误:WARNING: Sub-Space-Matrix is not hermitian in DA V 4 -4.681828688433112E-002对策:通过将默认AMIX=0.4,修改成AMIX=0.2(或0.3),问题得以解决。
以下是类似的错误:WARNING: Sub-Space-Matrix is not hermitian in rmm -3.00000000000000RMM: 22 -0.167633596124E+02 -0.57393E+00 -0.44312E-01 1326 0.221E+00BRMIX:very serious problems the old and the new charge density differ old charge density: 28.00003 new 28.06093 0.111E+00错误:WARNING: Sub-Space-Matrix is not hermitian in rmm -42.5000000000000ERROR FEXCP: supplied Exchange-correletion table is too small, maximal index : 4794错误:结构优化Bi2Te3时,log⽂件:WARNING in EDDIAG: sub space matrix is not hermitian 1 -0.199E+01RMM: 200 0.179366581305E+01 -0.10588E-01 -0.14220E+00 718 0.261E-01BRMIX: very serious problems the old and the new charge density differ old charge density: 56.00230 new 124.70394 66 F= 0.17936658E+01 E0= 0.18295246E+01 d E =0.557217E-02curvature: 0.00 expect dE= 0.000E+00 dE for cont linesearch 0.000E+00ZBRENT: fatal error in bracketingplease rerun with smaller EDIFF, or copy CONTCAR to POSCAR and continue但是,将CONTCAR拷贝成POSCAR,接着算静态没有报错,这样算出来的结果有问题吗?对策1:⽤这个CONTCAR拷贝成POSCAR重新做⼀次结构优化,看是否达到优化精度!对策2:⽤这个CONTCAR拷贝成POSCAR,并且修改EDIFF(⽬前参数EDIFF=1E-6),默认为10-4错误:WARNING: Sub-Space-Matrix is not hermitian in DA V 1 -7.626640664998020E-003⽹上参考解决⽅案:对策1:减⼩POTIM: IBRION=0,标准分⼦动⼒学模拟。

如何解决电脑内存频繁报错电脑内存频繁报错是使用电脑过程中常见的问题之一,可能会导致系统运行缓慢甚至崩溃。
针对这个问题,本文将介绍几种常见的解决方法,帮助解决电脑内存频繁报错的困扰。
一、清理不必要的后台程序和进程内存不足是电脑频繁报错的主要原因之一,因此清理不必要的后台程序和进程是解决该问题的首要步骤。
通过以下操作来实现:1. 打开任务管理器:按下“Ctrl”+“Shift”+“Esc”组合键,或者右键点击任务栏空白处选择“任务管理器”选项。
2. 切换到“进程”选项卡:在任务管理器中,点击“进程”选项卡,列出当前正在运行的所有进程。
3. 结束不必要的进程:在进程列表中,挑选那些你确定不再使用的进程,并右键点击选择“结束任务”选项。
二、增加系统内存如果清理后台程序和进程后问题仍未解决,那么可能是因为系统内存不足导致的。
考虑以下方式来增加系统内存:1. 添加物理内存:如果你的电脑内存插槽还有空余位置,可以购买适配的内存条并安装到插槽中,以增加系统的可用内存。
2. 虚拟内存设置:在没有物理内存插槽可用的情况下,可以通过设置虚拟内存来增加系统的内存容量。
三、更新或更换内存条电脑内存报错可能是由于内存条本身存在问题所致,所以检查并更新或更换内存条也是解决问题的一种方法。
具体步骤如下:1. 确定内存条插槽:打开电脑机箱,并找到内存条插槽的位置,记下每个内存条所在插槽的编号。
2. 检查内存条连接:小心地将内存条从插槽中取出,检查金手指接触是否良好,有无腐蚀或损坏。
3. 更换内存条:如果发现内存条存在问题,可以购买适配的内存条进行更换。
插入新的内存条时,确保与插槽对齐并轻轻按下,直到插槽两侧的扣子自动锁住。
四、运行内存诊断工具若以上方法未能解决问题,你还可以尝试运行内存诊断工具来发现和修复内存相关的错误。
下面是一些常用的内存诊断工具:1. Windows 内存诊断工具:在 Windows 操作系统中,你可以使用内置的内存诊断工具来检测和修复内存错误。
常见报错提示及处理办法
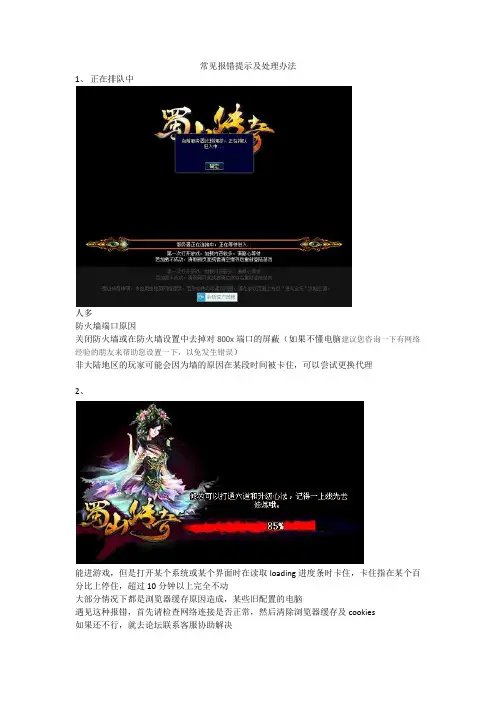
1、正在排队中
人多
防火墙端口原因
关闭防火墙或在防火墙设置中去掉对800x端口的屏蔽(如果不懂电脑建议您咨询一下有网络经验的朋友来帮助您设置一下,以免发生错误)
非大陆地区的玩家可能会因为墙的原因在某段时间被卡住,可以尝试更换代理
2、
能进游戏,但是打开某个系统或某个界面时在读取loading进度条时卡住,卡住指在某个百分比上停住,超过10分钟以上完全不动
大部分情况下都是浏览器缓存原因造成,某些旧配置的电脑
遇见这种报错,首先请检查网络连接是否正常,然后清除浏览器缓存及cookies
如果还不行,就去论坛联系客服协助解决
3、黑屏(具体表现为,游戏画面处显示黑色,其余部分,包括顶部导航,上方banner等都显示正常)
战斗
4、白屏(具体表现为,游戏画面处显示白色,其余部分,包括顶部导航,上方banner等都显示正常)
5、页面一片空白,上方导航条和都看不见
服务器维护中[2]。
6、flash崩溃
请关掉多余的进程和不必要的程序(如果不会可以使用360安全卫士清理内存)然后重新打开
7、
8、
9、
1。

如何解决电脑内存频繁报错的问题电脑内存频繁报错是许多电脑用户经常遇到的问题。
这个问题不仅会影响电脑的性能,还可能导致数据丢失和系统崩溃。
在本文中,我将分享一些解决电脑内存频繁报错问题的方法,帮助读者更好地应对这一挑战。
首先,我们需要了解什么是内存报错。
内存报错是指电脑内存出现问题,导致系统无法正常运行。
这种问题通常会导致蓝屏错误、应用程序崩溃或电脑变得非常缓慢。
虽然内存报错可能由多种原因引起,但最常见的原因是内存模块损坏或不兼容。
解决内存报错问题的第一步是检查内存模块。
您可以通过打开电脑机箱并检查内存模块是否正确安装来进行初步的检查。
如果发现内存模块松动或不正确插入,您可以尝试重新插入它们以解决问题。
如果内存模块没有问题,那么可能是模块本身损坏了。
在这种情况下,您可以尝试更换内存模块来解决问题。
除了检查内存模块,您还可以运行内存诊断工具来检测内存问题。
Windows操作系统提供了内置的内存诊断工具,可以帮助您检测和修复内存问题。
您可以通过按下Win + R键,然后输入“mdsched.exe”来运行内存诊断工具。
这将启动一个内存诊断工具的窗口,您可以选择在下一次重启时运行它。
在重启后,系统将自动运行内存诊断工具并报告任何内存问题。
如果内存报错问题仍然存在,那么可能是由于内存模块不兼容引起的。
在这种情况下,您可以尝试更换内存模块或添加新的内存模块来解决问题。
在购买新的内存模块时,确保选择与您的电脑兼容的型号和规格。
如果您不确定应该购买哪种内存模块,可以咨询专业人士或参考电脑制造商的建议。
除了硬件问题,软件问题也可能导致内存报错。
一些应用程序可能会占用过多的内存资源,导致系统出现错误。
为了解决这个问题,您可以尝试关闭一些不必要的应用程序或服务,以释放内存资源。
您还可以通过更新操作系统和应用程序来修复软件相关的问题。
更新通常包含修复程序和改进的兼容性,可以帮助您解决内存报错问题。
最后,定期清理电脑也是解决内存报错问题的重要步骤。

电脑报错提示解决方法大全电脑在使用过程中,经常会出现各种各样的错误提示。
这些错误提示可能是由软件问题、硬件故障或者系统错误引起的。
为了帮助大家更好地解决电脑报错问题,本文整理了一些常见的电脑报错提示以及对应的解决方法。
希望能够对大家有所帮助。
一、蓝屏错误提示1. 错误提示:STOP 0x0000001EKMODE_EXCEPTION_NOT_HANDLED解决方法:升级或者回滚显卡驱动程序,检查是否有硬件冲突,更新操作系统。
2. 错误提示:STOP 0x0000007ESYSTEM_THREAD_EXCEPTION_NOT_HANDLED解决方法:检查硬件兼容性,更新硬件驱动程序,查杀病毒。
3. 错误提示:STOP 0x000000D1DRIVER_IRQL_NOT_LESS_OR_EQUAL解决方法:更新或者卸载驱动程序,检查硬件是否损坏,关闭不必要的后台程序。
二、应用程序错误提示1. 错误提示:应用程序无法正常启动,xx.dll丢失解决方法:重新安装或者升级相关应用程序,修复系统文件。
2. 错误提示:该应用程序无法在当前计算机上执行解决方法:检查应用程序的系统要求,更新操作系统至满足要求的版本。
3. 错误提示:应用程序停止响应解决方法:关闭其他不必要的后台程序,清理系统垃圾文件,修复或者重新安装应用程序。
三、硬件错误提示1. 错误提示:硬盘SMART错误解决方法:备份数据,更换新的硬盘,及时进行数据恢复。
2. 错误提示:内存故障,请重新安装内存模块解决方法:重新安装内存模块,确保其插槽良好接触。
3. 错误提示:CPU温度过高,请立即关机解决方法:清理散热器,更换散热硅脂,确保电脑散热良好。
四、系统错误提示1. 错误提示:系统文件损坏,请运行系统修复工具解决方法:运行系统自带的修复工具,如Windows下的sfc/scannow命令。
2. 错误提示:磁盘空间不足解决方法:清理系统垃圾文件,删除不必要的文件和程序,扩展磁盘空间。

AE各种报错的原因和解决方案汇总AE(After Effects)是一款专业的视频处理软件,但在使用过程中,可能会遇到各种报错。
下面是一些常见的AE报错以及解决方案的汇总。
1. “After Effects错误:无法读取媒体文件”原因:可能是媒体文件路径错误、文件已被删除、文件格式不受支持等。
解决方案:-检查媒体文件路径是否正确,确认文件是否存在。
-尝试将文件重命名为不包含特殊字符或中文的名称。
-如果文件格式不受支持,可以尝试将文件转换为AE支持的格式。
2. “After Effects错误:分配更多内存、快速确定解决方案”原因:AE使用的内存超过了系统可用内存限制。
解决方案:-关闭一些占用大量内存的程序,释放系统内存。
3. “After Effects错误:无法创建足够的缓存大小”原因:AE需要创建较大的缓存,但系统可用内存不足。
解决方案:-关闭其他占用大量内存的程序。
-尝试通过增加计算机的物理内存来解决问题。
4. “After Effects错误:堆栈溢出”原因:AE脚本或插件出现了迭代次数过多的问题。
解决方案:-检查使用的脚本或插件是否与当前版本的AE兼容,并更新到最新版本。
-确保脚本或插件没有死循环的问题。
-尝试在执行脚本或插件之前减少项目中的复杂性。
5. “After Effects错误:无法初始化Mediacore”原因:可能是由于损坏的设备驱动程序、缺少插件或错误的设置引起的问题。
解决方案:-检查设备驱动程序是否最新,尝试更新驱动程序。
-确保所有必需的插件已正确安装,并尝试重新安装有关插件。
-检查AE的设置,确保所有设置正确。
6. “After Effects错误:无法为您呈现此帧”原因:可能是由于缺少所需的资源、过多的效果或高分辨率引起的问题。
解决方案:-确保所有所需的素材和资源都可用。
-禁用一些占用较多资源的效果。
-尝试减少分辨率或降低渲染质量。
总结来说,AE报错的原因通常包括文件丢失、系统资源不足、插件兼容性问题等。

python报错与解决错误类型总览IndexErrorAttributeErrorSyntaxErrorTypeErrorIndentationErrorNameErrorIOErrorKeyErrorIndexError- Python中的列表索引超出范围AttributeError- 当前对象类型并没有你调⽤的这⼀⽅法 / 属性,例如,对于 Python 3,你在⼀个 List 对象上调⽤ add ⽅法(列表没有 add ⽅法)SyntaxError- 忘记在字符串两端都加上单引号 '(或双引号 ")- 忘记在 def、if、for ⾏末添加冒号 :左括号与右括号数量不匹配,包括⼩括号 ()、中括号 [] 和⼤括号 {}TypeError- 在⼀个错误类型的对象上执⾏某个操作,例如,你拿⼀个 List 对象去除⼀个整型数字,或是想要改变⼀个 immutable 类型(如 Tuple, String)变量的值- 你认为有值的对象其实为 None- 使⽤⼀个⾮整型数字作为列表的索引- 在调⽤⼀个⽅法时,传⼊错误数量或错误类型的参数IndentationError- ⼀个代码块内的代码缩进了但没有对齐- 在代码缩进时混⽤了空格和制表位(有些⾏使⽤空格缩进,有些则使⽤制表位),虽然有时这些代码看上去是对齐了的NameError- 错拼了⼀个变量、函数或⽅法的名字- 忘记导⼊对应模块- 忘记定义该变量- 在函数定义前调⽤该函数- 在变量作⽤域之外使⽤该变量,例如,在 for 外使⽤⼀个在该 for 循环内部定义的⼀个变量- 在使⽤ print 输出单个字符时,忘记给该字符加上引号,例如,你只想输出字母 a,但写成了 print(a),这是程序会认为你要输出变量 a,但名为 a 的变量并未被定义IOError- 你试图打开⼀个不存在的⽂件KeyError- 你试图从⼀个字典中获取⼀个并不存在的 key 的值----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- ----- -----报错:ImportError: cannot import name 'xxxxx' from 'xxxx'据说:“ ⼀个是路径问题,⼀个是命名问题”根据⼀个实际案例,是换成低版本的库就可以使⽤,那估计是⽂件命名冲突的问题。

倍福AX5000伺服运行中报错FD43解决方案
1.检查硬件连接:首先,确保电源连接稳定,检查电缆连接是否正确。
使用示波器检查编码器或传感器的输出信号,并确认信号是否稳定和准确。
3.检查PID参数:PID参数对于伺服控制至关重要。
确保PID参数设
置正确,以保证伺服驱动器能够正常运行。
可以尝试使用默认的PID参数,并根据实际需求进行微调。
4.调整速度限制:如果伺服驱动器在高速运行时出现FD43错误,可
能是因为速度限制设置过低。
适当调整速度限制,确保在设定的运行速度
范围内正常运行。
5.验证反馈装置:检查编码器或其他反馈装置是否正常工作。
通过检
查反馈信号是否稳定,并对其进行校准,确保反馈装置能够提供准确的位
置和速度反馈。
6.更新固件版本:如果以上方法无法解决问题,尝试更新倍福
AX5000伺服驱动器的固件版本。
新版本的固件可能修复了原有版本的一
些问题。
内存报错怎么解决不知道你们有没有遇到内存报错的情况,不知道的话跟着店铺一起来学习了解内存报错怎么解决。
内存报错的解决方法1 内存条坏了更换内存条 (这个可能性比较小,一般来说在XP系统下的内存如果是坏了,基本状态是突然重启)2 双内存不兼容使用同品牌的内存或只要一条内存 (这个有一定的可能性,曾经也插过两个不同品牌的内存在一台机器上,当时如果没有出现不兼容的症状,以后的使用中也没什么问题)3 内存质量问题更换内存条(有可能,个人来说,根本无法测试内存是否质量有问题)4 散热问题加强机箱内部的散热(可能性比较大,散热不好经常会导致软件使用错误)5 内存和主板没插好或其他硬件不兼容重插内存或换个插槽(内存没插好的可能性比较小,我的经验来看内存如果没插好,通常会出现,无法启动的状态)6 硬件有问题更换硬盘(这个意思就是,有可能在读写虚拟内存的时候出现错误,硬盘上有坏道,虚拟内存有可能读写在坏道上,就导致错误提示)7~12都是软件的兼容性问题造成的内存报错,都具有一定的可能性.7 驱动问题重装驱动,如果是新系统,应先安装主板驱动(有可能,装的驱动不好,会造成兼容性的问题)8 软件损坏重装软件9 软件有BUG 打补丁或更新到最新版本10 软件和系统不兼容给软件打上补丁或是试试系统的兼容模式11 软件和软件之间有冲突如果最近安装了什么新软件,卸载了试试12 软件要使用其他相关的软件有问题重装相关软件,比如播放某一格式的文件时出错,可能是这个文件的解码器有问题13 病毒问题杀毒14 杀毒软件与系统或软件相冲突由于杀毒软件是进入底层监控系统的,可能与一些软件相冲突,卸载试试15 系统本身有问题有时候操作系统本身也会有BUG,要注意安装官方发行的更新程序,象SP的补丁,最好打上.如果还不行,重装系统,或更换其他版本的系统。
另外的说法还有:在控制面板的添加/删除程序中看看你是否安装了微软NET.Framework,如果已经安装了,可以考虑卸载它,当然如果你以后在其它程序需要NET.Framework时候,可以再重新安装。
keil编译常见报错和解决方法Keil是一款常用的嵌入式开发工具,用于编写和调试嵌入式系统的程序。
在使用Keil编译时,我们常常会遇到一些报错信息。
本文将介绍一些常见的Keil编译报错及解决方法,帮助读者快速解决问题。
1. "Error: L6218E: Undefined symbol"错误这个错误通常是由于使用了未定义的变量或函数导致的。
解决方法是检查代码中使用的符号是否正确定义或是否包含了正确的头文件。
如果符号确实未定义,需要在代码中进行定义或者引入相关的头文件。
2. "Error: L6002U: Could not open file"错误这个错误表示编译器无法打开指定的文件。
解决方法是检查文件路径是否正确,文件是否存在,并且是否具有读取权限。
如果文件路径正确但是依然无法打开,可能是文件被其他程序占用或者权限设置不正确,需要解决这些问题后重新编译。
3. "Error: C2513: 'function' : no variable declared before '=' "错误这个错误表示在赋值语句中使用了未声明的变量。
解决方法是检查变量是否正确声明,并确保在赋值之前进行了声明。
如果变量确实未声明,需要在代码中添加相应的变量声明。
4. "Error: C2065: 'variable' : undeclared identifier"错误这个错误表示使用了未声明的变量。
解决方法是检查变量是否正确声明,并确保在使用之前进行了声明。
如果变量确实未声明,需要在代码中添加相应的变量声明。
5. "Error: C1083: Cannot open include file: 'header.h': No such file or directory"错误这个错误表示编译器无法找到指定的头文件。
LR常见报错及解决的办法LR常见报错及解决的办法1、LoadRunner 26377、26388错误码的成因脚本如下:脚本的是请求下载,如果是三个cot请求,就不会有错,我现在是有10个cot请求,从"objectURI4"就提示以下错误信息,请大有帮忙解决一下。
谢谢错误码如下:Error -26377: No match found for the requested parameter "objectURI10". Check whether the requested boundaries exist in the response data. Also, if the data you want to save exceeds 1516000 bytes, use web_set_max_html_param_len to increase the parameter size [MsgId: MERR-26377]web_url("entry") highest severity level was "ERROR", 1631 body bytes, 199 header bytes [MsgId: MMSG-26388]1.首先看下脚本中有没有使用了自动关联(web_reg_save_param)2.在Virtual的脚本里查询下web_reg_save_param的参数使用位置,然后把这个参数化给还原回来,比如web_reg_save_param("Siebel_Analytic_ViewState2",............然后就在全文查询Siebel_Analytic_ViewState23,至于修改成什么东西要看几个地方,如果是启动了自动关联,一般在脚本上面会有一段被自动注释掉的:关联变量名="值"比如上面的Siebel_Analytic_ViewState2大概就是// {Siebel_Analytic_ViewState2}="/wEPDwUKMTI5Nzk1OT c3NmRkikSkNLllgC5BL8dbmU5bHIwtt4 I="那么这里的/wEPDwUKMTI5Nzk1OTc3NmRkikSkNLllgC5BL8dbmU5bHIwtt4I =就是要找的值了,这个也可以在"View Tree"里找到4.把"View script"里的被关联的那部分参数改成/wEPDwUKMTI5Nzk1OTc3NmRkikSkNLllgC5BL8dbmU5bHI wtt4I=就好了(不是修改web_reg_save_param里的参数,要把它注释掉,从下面正文里查询另一个带Siebel_Analytic_ViewState2的东东,把它改掉)把web_set_max_html_param_len(50000)值加大这个函数要放在所有参数化前面。
报错4013的解决方法一、了解4013报错。
1.1 首先呢,这个4013报错啊,就像一个突然冒出来的小怪兽,挡在我们顺利做事的道路上。
它可不是个小麻烦,很多朋友遇到它就头疼得很。
这个报错通常在一些设备或者软件运行的时候出现,比如说手机刷机或者更新系统的时候。
1.2 一般情况下,4013报错就表示设备和服务器之间的通信出了问题。
就好比你跟朋友打电话,结果线路断了,两边都听不见对方说啥了。
这时候设备不知道该怎么继续进行操作,就只能给你报个4013错误,告诉你事情没办成。
二、可能的原因。
2.1 硬件连接问题。
这是很常见的一个原因。
就像盖房子,地基没打好,楼肯定盖不起来。
如果你的设备和电脑之间的连接线有问题,比如说数据线损坏了,那就像断了的桥梁,数据过不去,就容易出现4013报错。
有时候接口松动也会这样,就像插头没插紧,电都通不了,数据更没法传输了。
2.2 软件故障。
软件有时候也会调皮捣蛋。
可能是你正在使用的软件版本有漏洞,就像衣服破了个洞,不补好就会漏风。
或者是软件的某些设置不对,就像你把闹钟设置错了时间,它就不能在正确的时候响。
比如说软件的权限设置,如果没有给到足够的权限让它去和服务器通信,那肯定就会报错4013啦。
2.3 网络问题。
网络就像信息的高速公路。
如果网络不稳定,那数据传输就会像在坑坑洼洼的路上开车一样,颠颠簸簸的。
要是网络中断了,那就更糟糕了,就像高速公路突然塌方了,数据根本就过不去,4013报错就很可能出现了。
比如说你在信号不好的地方进行设备更新,那遇到这个报错的几率就大大增加了。
三、解决方法。
3.1 检查硬件。
先把数据线拔下来,好好看看有没有破损的地方。
要是有,那就赶紧换一根新的数据线,这就像给断了的桥梁重新搭一座一样。
然后把接口也检查一下,确保插得紧紧的,别让它松松垮垮的。
这就好比把插头插稳,电才能通得顺畅。
3.2 处理软件。
如果是软件版本的问题,那就去看看有没有新的版本可以更新。
新⼿常见6种的python报错及解决⽅法此篇⽂章整理新⼿编写代码常见的⼀些错误,有些错误是粗⼼的错误,但对于新⼿⽽已,会折腾很长时间才搞定,所以在此总结下我遇到的⼀些问题。
希望帮助到刚⼊门的朋友们。
Error变量名错误报错:>>> print aTraceback (most recent call last):File "<stdin>", line 1, in <module>NameError: name 'a' is not defined解决⽅案:先要给a赋值。
才能使⽤它。
在实际编写代码过程中,报NameError错误时,查看该变量是否赋值,或者是否有⼤⼩写不⼀致错误,或者说不⼩⼼将变量名写错了。
注:在Python中,⽆需显⽰变量声明语句,变量在第⼀次被赋值时⾃动声明。
>>> a=1>>> print a12.IndentationError代码缩进错误点击返回⽬录代码:a=1b=2if a<b:print a报错:IndentationError: expected an indented block原因:缩进有误,python的缩进⾮常严格,⾏⾸多个空格,少个空格都会报错。
这是新⼿常犯的⼀个错误,由于不熟悉python编码规则。
像def,class,if,for,while等代码块都需要缩进。
缩进为四个空格宽度,需要说明⼀点,不同的⽂本编辑器中制表符(tab键)代表的空格宽度不⼀,如果代码需要跨平台或跨编辑器读写,建议不要使⽤制表符。
解决⽅案:a=1b=2if a<b:print a3.AttributeError对象属性错误报错:>>> import sys>>> sys.PathTraceback (most recent call last):File "<stdin>", line 1, in <module>AttributeError: 'module' object has no attribute 'Path'原因:sys模块没有Path属性。
设置:初始值收敛值结果AMIX =0.0100;BMIX =0.0001 AMIX = 0.01; BMIX = 0.00 计算无误AMIX = 0.1000;BMIX = 0.0010 AMIX = 0.10; BMIX = 0.00 计算无误AMIX =0.20; BMIX = 0.01 AMIX =0.20; BMIX = 0.01 计算无误AMIX=0.2、BMIX=0.001 AMIX=0.2、BMIX=0.001 计算无误AMIX=0.3、BMIX=0.1 AMIX=0.3、BMIX=0.1 计算无误AMIX=0.4 AMIX = 0.40; BMIX = 1.00 静态log: WARNING in EDDRMM: call toZHEGV failed, returncode = 6 3 **,能带一样AMIX=0.02 AMIX = 0.02; BMIX = 1.00 计算无误AMIX=0.1 AMIX = 0.10; BMIX = 1.00 静态log: WARNING in EDDRMM: call toZHEGV failed, returncode = 6 3 **,能带一样AMIX=0.3 AMIX = 0.30; BMIX = 1.00 静态log: WARNING in EDDRMM: call toZHEGV failed, returncode = 6 3 **,能带一样BMIX=0.0001 AMIX = 0.40; BMIX = 0.00 计算无误以上参数设置,得到的能带图都一样,如下图:综上:设置AMIX=0.2(或0.3),BMIX默认(省事,等于1.0),可以保证计算过程无误。
还需进一步调整其他参数,算出正确的能带。
警告:算1QL弛豫、静态、能带时,都有这个提示:ADVICE TO THIS USER RUNNING 'V ASP/V AMP' (HEAR YOUR MASTER'S VOICE ...): You have a (more or less)'small supercell' and for smaller cells it is recommended to use the reciprocal-space projection scheme! The real space optimization is not efficient for small cells and it is also less accurate ... Therefore set LREAL=.FALSE. in the INCAR file对策:对于较小的晶胞(原子数小于20),设置LREAL=.FALSE.,计算结果比较精确。
而对于较大的晶胞,设置LREAL=Auto,这样计算速度比较快。
本体系含原子5个,INCAR中LREAL=Auto。
设置所有INCAR中的LREAL=.FALSE.,重新算一遍。
对于1QL 2QL 3QL原子数分别为5、10、15,LREAL=.False.对于4QL 5QL 6QL原子数分别为20、25、30,LREAL=Auto自旋轨道耦合计算时,静态和能带计算中出现的错误:ERROR: non collinear calculations require that V ASP is compiled without the flag -DNGXhalf and -DNGZhalf分析:V ASP手册中关于自旋轨道耦合计算的描述(翻译版):•【SAXIS =自旋轴方向;MAGMOM= 每个原子的初始磁矩值】2) 不要忘记to include SOC, please1) add the following lines to INCARLNONCOLLINEAR=.True.LSORBIT=.True.SAXIS = # please give the spin quantization axis here, like 0 0 1 for the z-axis)MAGMOM= # please give a triplet of numbers for each atom here, and please have a look at the manual (chapter non-collinear calculations and spin-orbit tag) on how the direction of the magnetic moments has to be defined with respect to the spin-quantization axis)LORBMOM=.True.ISYM= -12)不要忘记如果你用的vasp不包含任何预编译程序命令-DNGXhalf, -DNGZhalf, -DwNGXhalf, -DwNGZhalf ,你必须重新编译vasp,因为这些参数通常对于非线性磁性计算是必要的,在DOSCAR中的第二块数据包含了E和4列s,p,d,如下:rho, m_x, m_y, m_z ,2) don't forget that you may have to re-compile vasp without any of the precompiler (CPP) flags set: -DNGXhalf, -DNGZhalf, -DwNGXhalf, -DwNGZhalf , as necessary for non-collinear runs in general for non-collinear magnetism, the second block of data in DOSCAR contains E, and 4 columns for each, s,p,d, giving:rho, m_x, m_y, m_zwith m....magnetisation,it makes absolutely NO SENSE to set ISPIN=2 (up and down) for non-clollinear runs, therfore this tag is ignored when it s read from INCAR.Symbol DescriptionΓ Center of the Brillouin zoneSimple cubeM Center of an edgeR Corner pointX Center of a faceFace-centered cubicK Middle of an edge joining two hexagonal facesL Center of a hexagonal faceU Middle of an edge joining a hexagonal and a square faceW Corner pointX Center of a square faceBody-centered cubicH Corner point joining four edgesN Center of a faceP Corner point joining three edgesHexagonalA Center of a hexagonal faceH Corner pointK Middle of an edge joining two rectangular facesL Middle of an edge joining a hexagonal and a rectangular faceM Center of a rectangular face1) it does not look to me as if the magnetic convergence is particularly bad. (please dont compare the moments stemming fromthe augmentation to the total moments).have you decreased AMIX,BMIX, AMIX_MAG and BMIX_MAG for this run?2)the mixing parameters must not have any influence on the converged total energies.3) if your system has a magnetic moment, you have to set ISPIN.unless you set LNONCOLLINEAR explicitely , collinear magnetism is assumed by default, there is nothing to be specified in extra (except from starting with FM or AFM configuration by choosing the MAGMOMs accordingly)4) please in any case check if the convergence of ALL ionic steps is bad. (consider that it may be possible that you relaxed into an unreasonable geometry which does not converge electronically).without knowing further details, I would recommend to try the following:please keep the low mixing parameters check if the k-mesh is converged try if a different BZ-integration (ISMEAR=1) and slightly larger smearing (SIGMA) helps set LMAXMIX=6 if your system contains d-elementsISYM-tag and SYMPREC-tagISYM = 0|1|2|3Default 1switch symmetry on (1, 2 or 3) or off (0).For ISYM=2 a more efficient, memory conserving symmetrisation of the charge density is used. This reduces memory requirements in particular for the parallel version. ISYM=2 is the default if PAW data sets are used.ISYM=1 is the default if VASP runs with US-PP’s.For ISYM=3, the forces and the stress tensor only are symmetrized, whereas the charge density is left unsymmetrized (VASP.5.1 only).This option might be useful in special cases, where charge/orbital ordering lowers the crystal symmetry, and the user wants to conserve 【保存, 保藏】the symmetry of the positions during relaxation.However, the flag must be used with great caution, since a lower symmetry due to charge/orbital ordering, in principle also requires to sample the Brillouin zone usinga k-point mesh compatible with the lower symmetry caused by charge/orbital ordering.The program determines automatically the point group symmetry and the space group according to the POSCAR file and the line MAGMOM in the INCAR file.The SYMPREC-tag (VASP.4.4.4 and newer versions only) determines how accurate the positions in the POSCAR file must be. The default is 10−5, which is usually suffiently large even if the POSCAR file has been generated with a single precisionprogram.Increasing the SYMPREC tag means, that the positions in the POSCAR file can be less accurate.During the symmetry analysis, VASP determines• the Bravais lattice type of the supercell,• the point group symmetry and the space group of the supercell wit h basis (static and dynamic) - and prints the namesof the group (space group: only ’family’),• the type of the generating elementary (primitive) cell if the supercell is a non-primitive cell,• all ’trivial non-trivial’ translations (= trivial translatio ns of the generating elementary cell within the supercell) —needed for symmetrisation of the charge,• the symmetry-irreducible set of k-points if automatic k-mesh generation was usedand additionally the symmetry irreducible set of tetrahedra if the tetrahedron method was chosen together with the automatic k-mesh generation and of course also the corresponding weights (’symmetry degeneracy’),• and tables marking and connecting symmetry equivalent ions.The symmetry analyses is done in four steps:• First the point group symmetry of the lattice (as supplied by the user) is determined.• Then tests are performed, whether the basis breaks symmetry. Accordingly these symmetry operations are removed.• The initial velocities are checked for symmetry breaking.• Finally, it is checked wheter MAGMOM breaks the symmetry. Correspondingly themagnetic symmetry group is determined (VASP.4.4.4 and newer releases only; if you use older version please also see section 6.12). The program symmetrises automatically:• The t otal charge density according to the determined space group• The forces on the ions according to the determined space group.• The stress tensor according to the determined space groupWhy is symmetrisation necessary: Within LDA the symmetry of the supercell and the charge density are always the same.This symmetry is broken, because a symmetry-irreducible set of k-points is used for the calculation.。