5_ProteinPurification(1)
- 格式:ppt
- 大小:15.29 MB
- 文档页数:12

Protein Expression and Purification22,159–164(2001)160SCOTT A.LESLEYTABLE1Genomic versus Proteomic TechnologiesGenomic technologies(DNA)Proteomic technologies(protein) Identification Determined experimentally,bioinformatics Predicted from genomic informationFunction1-dimensional information storage3-dimensional organization of chemicalfunctionalitiesBuilding blocks4bases20ϩamino acidsDetection sensitivity PCR amplification techniques Direct detection methodsSynthetic approaches Cheap and efficient oligonucleotide synthesis Limited capacity of peptide synthesismethods combined with PCRSequence determination500–700bases common by automated sequencing Direct sequencing difficult,mass spectrometry Purification Generic methods Generic methods require modification of proteinthrough gene fusionAnalysis methods Typically employ enzymes,hybridization Chemical,biophysical,biochemicaland mutagenesis),interactions between proteins(two-are of primary interest and typically are expressed in hybrid),and global protein changes(2D gels and LC–a bacterial host.Often this approach leads to problems MS).Purified protein is often required in these studies associated with expression levels and proper folding of and defines the outputs of any parallel expression and the protein of interest.Flexibility in expression options purification process.is a key parameter.Pichia or baculovirus expressionsystems can offer effective alternatives to bacterial sys-tems.Each expression scenario requires a specific vec-GENE CLONING FOR EXPRESSIONtor.Recloning cDNAs into each of these specific vectors Determining gene function through genomics typi-is extremely labor-intensive.Recombinatorial cloning cally starts from a query of a database.Sequence infor-methods provide an opportunity to minimize the effort mation for the3.9billion bases of sequence from the required for alternate expression.human genome is now available(1,2).Access and inter-Two systems are commonly used for recombinatorial pretation of this information often require sophisticated cloning and shown in Fig.1.The cre–lox recombination bioinformatics software outside the scope of this dis-system described by Elledge utilizes a single recombina-cussion.Public archives such as the Unigene Data-tion to introduce the gene of interest into a recipient base(/)or TIGR(http://vector(3).This is an in vitro reaction combined with a /)provide bioinformatic access to many in-genetic selection for the recombinant vector.In this way teresting plete genomic sequence infor-the gene of interest is cloned once into a donor vector mation is now available through Celera(http://and can then be moved into any of a number of recipient /index.cfm).Future annotation of fullplasmids for expression in different hosts or to utilize sequence information will greatly expand the access todifferent purification tags.A similar system utilizes full-length cDNA sequences.The first requirement is converting this genomic in-lambda Int/Xis/IHF recombination at att sites(4)to formation into an actual cDNA clone of that gene.Am-achieve transfer of open reading frames(ORFs).This plification of full-length cDNAs via PCR is the typical system has the advantage of a precise ORF transfer to first step.Both reverse transcriptase and amplification the expression vector rather than the cointegrant vector polymerases typically are lacking in proofreading activ-product of the cre–lox system.With either system,the ity.Care must be taken to limit the number of steps of primary limitation is that translational fusion of the amplification and to use proofreading enzymes where recombination sites is typically required to maintain possible to minimize the probability of introducing un-the flexibility and utility of the recombination method wanted mutations.Tissue selection for cDNA librariesfor expression.In those cases,such as crystallography, also is an important consideration for attempting towhere translational fusions are potentially detrimental isolate genes as they must be expressed within thatto the protein,a conventional cloning approach is library source for successful amplification.This infor-more appropriate.mation is often obtained from cDNA or oligonucleotideRegardless of the cloning method,parallel expression expression arrays.and purification requires utilization of purification Amplified gene products are cloned into appropriatetags.Many options exist for this purpose.A comprehen-vectors for expression.Depending on the source of thesive review is beyond the scope of this article.A list of gene,the host,and the end use of the protein,manydifferent vectors may be appropriate.Eukaryotic genes some commonly used tags is shown in Table2.By farHIGH-THROUGHPUT PROTEOMICS161FIG.1.Strategies of recombinatorial cloning.Individual cDNAs are cloned into a donor vector that can then be recombined into any number of recipient vectors through recombinatorial cloning.One option is to form a cointegrant plasmid through Cre-mediated recombination across a lox site.In the second scenario,a cDNA is flanked by phage lambda att sites which direct recombination into an expression vector through the use of the INT/XIS/IHF proteins.In these ways,a single donor clone can easily be transferred into any number of recipient vectors. the most common fusion is the histidine tag for purifica-analysis studies into small-scale for analysis of expres-tion on metal-chelate resins.This tag provides a sub-sion levels and properties and into large-scale for use stantial purification handle while being relatively un-with many of the proteomic applications.obtrusive as a fusion partner.Beyond purification,Small-scale expression is most useful for identifying translational fusions often provide a means to enhance those clones which express recombinant protein to high expression.The larger fusion tags such as thioredoxin levels and for evaluating the folding state of the protein. and GST often are superior in this respect.Crude expression testing is typically done by simpleSDS–PAGE analysis of whole cells.Evaluation of thefolding state is typically done by centrifugal fraction-RECOMBINANT EXPRESSION OF PROTEINation of a lysate,requiring a more gentle lysis proce-dure.Several lysozyme and mild detergent methods are Bacterial expression is most common for recombinantcommercially available for this purpose.proteins because of its ease of use and the high levelsof protein obtained.It is useful to divide expression For large-scale production for many applications,TABLE2Common Purification TagsBasis of purification Elution Reference Small tagsHistidine tag Metal affinity resin Imidizole(5)S-tag Interaction with S-protein Temperature(6) Calmodulin-binding protein Interaction with Calmodulin Calcium(7) Large tagsGlutathione S-transferase Glutathione agarose Reduced glutathione(8) Thioredoxin Phenylarsine oxide resin-Mercaptoethanol(9) Biotinylation domain Monomeric avidin resin Biotin(10) Maltose binding protein Amylose resin Maltose(11) Chitin binding protein/intein Chitin affinity Thiol(12)162SCOTT A.LESLEYtens of milligrams of protein typically are required.PURIFICATION STRATEGYEven with common bacterial expression levels,500–Proteins are highly diverse in their properties mak-1000ml of culture typically is required to provide these ing generic methods of purification difficult.Purifica-amounts.While such methods are commonplace in labo-tion tags such as described in Table2are a typical ratories,systems for parallel processing large numbers solution for purifying proteins in parallel.His-tag fu-of cultures at this level are not commercially available.sions are very common and provide a single-step chro-By developing instrumentation and optimizing media matographic purification that yields protein of suffi-and aeration conditions for high-density cell growth,cient purity for most applications.In addition,the his-our laboratory can parallel process96cultures at this tag sequence requires the addition of only six amino scale.Optical density(OD)values at600nm can reach acids to the recombinant protein,reducing the likeli-a value of40with logarithmic growth through at least hood that such a fusion will adversely affect gene func-30OD units.These cell densities allow us to producetion.A typical purification strategy is outlined in Fig.2. sufficient cell mass with65-ml culture volumes to yieldtens of milligrams of recombinant protein,sufficient for PURIFICATION AUTOMATIONmost applications.Such instrumentation is not com-Parallel processing typically involves instrumenta-monly available,but common shaking incubators cantion for automation.Lysis methods such as sonication or substitute with larger volumes.using a French press are not simple automation tasks. Recombinant expression of proteins is achievedLikewise,centrifugation is not easily integrated into through induction of a strong promoter system.Manyautomation due to problems of locating and indexing options exist in this regard including tac,T7,lambdathe rotor position.Automation of this process typically P L,and ara B promoters.It is important for parallelinvolves protocol modifications.This can easily be processing that growth and induction characteristicsachieved in small-scale methods.are consistent.For this reason,it is important to retainOn small-scale,parallel processing usually involves tight repression of expression and have a simple induc-use of a96-well plate format.Lysis is typically achieved tion procedure for high-level expression.For T7sys-using a combination of lysozyme and freeze–thaw cy-tems,the lac operator and T7lysozyme(pLysS)provide cles.Phage lysozymes are more effective than hen egg an extra level of repression.The arabinose promoter is white lysozyme for this purpose and can be combined tightly repressed in the absence of inducer and is our with nucleases to reduce viscosity and facilitate re-preferred system for parallel growth.With all of the moval of cell debris at the low g forces commonly used promoters listed,recombinant expression levels of10–with microtiter plates.Alternatively,nonionic deter-50%of total cell protein are common.gents can be employed for nondenaturing lysis.FIG.2.Generalized purification strategy of recombinant fusion protein.A common purification strategy is shown here.Proteins are purified from fermentation cultures by affinity purification.Isolated cell pellets are resuspended in an appropriate lysis buffer and disrupted by high-intensity sonication.Cell walls and insoluble debris are pelleted by centrifugation and the soluble supernatant containing the recombinant fusion protein is applied to chromatography resin containing an immobilized metal for affinity purification.Fractions containing the recombinant protein can be used directly or further purified using conventional chromatographic techniques.HIGH-THROUGHPUT PROTEOMICS163TABLE3Robotic Systems with Capabilities Adaptable to ProteinPurificationManu-facturer Instrument WebsiteQiagen BioRobot3000/Tomtec Quadra96/Matrix PlateMate /Hamilton Microlab4200/Beckman Biomek2000/Packard PlateTrak /index2.htmGilson Nebula215/index.htmlRobotic systems for nucleic acid purification are rela-tively commonplace and have recently been adapted forprotein purification.The Qiagen BioRobot3000per-forms multiple functions relevant to protein purifica-tion.It provides aspirate,dispense,pipet,vacuum fil-tration,and plate-shaking functions on a relativelycompact platform.These functions can be adapted to FIG.4.SDS–PAGE analysis of purified protein.Metal affinity chro-perform cell lysis and chromatography steps from1–2matography yields highly purified protein from a single chromato-graphic separation.This gel shows typical yields and purity obtained ml of bacterial culture.Specialized96-well plates clearfrom parallel purification using an automated purification system. cell debris via vacuum filtration and are also used toSuch proteins have been incorporated directly in successful crystalli-retain resin for chromatographic separations.The Wal-zation trials.Ten-microliter samples of12ml protein eluates from Ni-lac Quadra96also has most of these capacities and resin were separated by10%SDS–PAGE.Samples are recombinant can parallel process96or384samples.Both of thesefusions of thioredoxin to human proteins as indicated by accessionnumber.systems have been used with success in our laboratoryfor small-scale protein purification of proteins in micro-titer plates.Table3lists some robotic systems that maybe applied to small-scale protein purification.providing the throughput needed for proteomic studies Despite the difficulties,large-scale protein purifica-involving tens of thousands of proteins.We are cur-tion also can be automated.In our laboratory we simul-rently able to process approximately96–192proteins taneously process96bacterial cultures of65–70ml.per day with this system with average yields of around Instrumentation for processing96parallel cultures at10mg of purified protein.Affinity purification results that scale required development of custom roboticsin recombinant protein that is typically80–90%pure shown in Fig.3.These robotics incorporate liquid aspi-(see Fig.4)which is sufficient for most applications. rate and dispense,centrifugation,and sonication capa-Subsequent purification is sometimes necessary,for ex-bilities required for purification.Automation is key to ample,in protein crystallography,and is achieved usingFIG.3.Protein purification automation.Custom robotics for performing the purification strategy outlined in Fig.2are shown.(a)The instrument has capacity for automated liquid aspiration and dispensing,sonication,centrifugation,and fractionation.Ninety-six cultures are processed in parallel,giving up to10–50mg of purified protein per culture.(b)Expanded view of aspirate/dispense/sonicate head accessing rotor.164SCOTT A.LESLEYREFERENCESstandard ion-exchange and size-exclusion chromatog-raphy.Automation of these methods is relatively1.Venter,J.C.et al.(2001)The sequence of the human genome. straightforward employing standard FPLC and au-Science291,1304–1351.tosampler instrumentation.2.International Human Genome Sequencing Consortium(2001)Initial sequencing and analysis of the human genome.Nature SUMMARY409,860–921.3.Liu,Q.,Li,M.Z.,Leibham,D.,Cortez,D.,and Elledge,S.J. Determining gene function and understanding the(1999)The univector plasmid-fusion system,a method for rapid relationships and interactions of the gene products are construction of recombinant DNA without restriction enzymes.a global effort in biological studies.The approach toCurr.Biol.8,1300–1309.performing this immense task is driven by the availabil- 4.Hatley,J.L.,Temple,G.F.,and Brasch,M.A.(2000)DNA cloningity of genomic information.To utilize this informationusing in vivo site-specific recombination.Genome Res.10,pp.1788–1795.for experimentation,however,significant effort isneeded to actually isolate and express proteins from5.Petty,K.J.(1996)Metal-chelate affinity chromatography,in“Current Protocols in Molecular Biology,”Vol.2,Wiley,New York. the genes of interest for study.The complexity of this6.Kim,J.S.,and Raines,R.T.(1993)Ribonuclease S-peptide as a effort is compounded by the large number of gene prod-carrier in fusion proteins.Protein Sci.2,348–356.ucts comprising the proteome.Parallel processing and7.Stofko-Hahn,R.E.,.Carr,D.W,and Scott,J.D.(1992)A single generic methods are required to achieve a systematicstep purification for recombinant proteins.FEBS Lett.302, and thorough evaluation of gene function.274–278.Experimental uses of proteins for structural and8.Smith,D.B.,and Johnson,K.S.(1988)Single-step purification functional studies typically require milligram amounts of polypeptides expressed in Escherichia coli as fusions within purified form.Unlike genomic technologies that pri-glutathione S-transferase.Gene67,31–40.marily involve the study of nucleic acids,proteomic9.Lu,Z.,DiBlasio-Smith,E.A.,Grant,K.L.,Warne,N.W.,LaVallie,studies focus on proteins.Proteins are by nature much E.R.,Collins-Racie,L.A.,Follettie,M.T.,Williamson,M.J.,more diverse in composition and properties than nucleicand McCoy,J.M.(1996)Histidine patch thioredoxins.Mutantforms of thioredoxin with metal chelating affinity that provide acids.In many ways,this makes them more interestingfor convenient purifications of thioredoxin fusion proteins.J. but also less amenable to generic methods and technolo-Biol.Chem.271,5059–5065.gies for parallel processing.Nonetheless,methods and10.Cronan,J.E.(1990)Biotination of proteins in vivo.A post-trans-instrumentation are currently available to meet this lational modification to label,purify,and study proteins.J.Biol.ing these advances will allow a systematic Chem.265,10327–33.effort at understanding biological pathways at the11.Maina,C.V.,Riggs,P.D.,Grandea,A.G.,Slatko,B.E.,Moran,molecular level.L.S.,Tagliamonte,J.A.,McReynolds,L.A.,and Guan,C.D.(1988)An Escherichia coli vector to express and purify foreign ACKNOWLEDGMENTS proteins by fusion to and separation from maltose-binding pro-tein.Gene74,365–373.The author acknowledges the help of Marc Nasoff,Heath Klock,Dan McMullan,Tanya Shin,Juli Vincent,Mike Hornsby,Mark12.Chong S.,Mersha F.B.,Comb D.G.,Scott M.E.,Landry D.,Vence L.M.,Perler F.B.,Benner J.,Kucera R.B.,Hirvonen C. Knuth,Loren Miraglia,and Jeremiah Gilmore for their contributionsto the high-throughput cloning and expression efforts.He also recog- A.,Pelletier J.J.,Paulus H.,and Xu M.Q.(1997)Single-column nizes Bob Downs,Mark Weselak,Andy Meyer,and Jim Mainquistpurification of free recombinant proteins using a self-cleavable and the rest of the GNF engineering staff for their contributions to affinity tag derived from a protein splicing element.Gene192, the custom robotics that make this effort possible.271–281.。

Instruction ManualProBond TM Purification SystemFor purification of polyhistidine-containing recombinant proteinsCatalog nos. K850-01, K851-01, K852-01, K853-01, K854-01,R801-01, R801-15Version K2 September200425-0006iiTable of ContentsKit Contents and Storage (iv)Accessory Products (vi)Introduction (1)Overview (1)Methods (2)Preparing Cell Lysates (2)Purification Procedure—Native Conditions (7)Purification Procedure—Denaturing Conditions (11)Purification Procedure—Hybrid Conditions (13)Troubleshooting (15)Appendix (17)Additional Protocols (17)Recipes (18)Frequently Asked Questions (21)References (22)Technical Service (23)iiiKit Contents and StorageTypes of Products This manual is supplied with the following products:Product CatalogNo.ProBond™ Purification System K850-01ProBond™ Purification System with Antibodywith Anti-Xpress™ Antibody K851-01with Anti-myc-HRP Antibody K852-01with Anti-His(C-term)-HRP Antibody K853-01with Anti-V5-HRP Antibody K854-01ProBond™ Nickel-Chelating Resin (50 ml) R801-01ProBond™ Nickel Chelating Resin (150 ml) R801-15ProBond™Purification System Components The ProBond™ Purification System includes enough resin, reagents, and columns for six purifications. The components are listed below. See next page for resin specifications.Component Composition Quantity ProBond™ Resin 50% slurry in 20% ethanol 12 ml5X NativePurification Buffer250 mM NaH2PO4, pH 8.02.5 M NaCl1 × 125 ml bottleGuanidinium LysisBuffer6 M Guanidine HCl20 mM sodium phosphate, pH 7.8500 mM NaCl1 × 60 ml bottleDenaturingBinding Buffer8 M Urea20 mM sodium phosphate, pH 7.8500 mM NaCl2 × 125 ml bottlesDenaturing WashBuffer8 M Urea20 mM sodium phosphate, pH 6.0500 mM NaCl2 × 125 ml bottlesDenaturing ElutionBuffer8 M Urea20 mM NaH2PO4, pH 4.0500 mM NaCl1 × 60 ml bottle3 M Imidazole,20 mM sodium phosphate, pH 6.0500 mM NaCl1 × 8 ml bottlePurificationColumns10 ml columns 6Continued on next pageivKit Contents and Storage, ContinuedProBond™Purification System with Antibody The ProBond™ Purification System with Antibody includes resin, reagents, and columns as described for the ProBond™ Purification System (previous page) and 50 µl of the appropriate purified mouse monoclonal antibody. Sufficient reagents are included to perform six purifications and 25 Western blots with the antibody.For more details on the antibody specificity, subclass, and protocols for using the antibody, refer to the antibody manual supplied with the system.Storage Store ProBond™ resin at +4°C. Store buffer and columns at room temperature.Store the antibody at 4°C. Avoid repeated freezing and thawing of theantibody as it may result in loss of activity.The product is guaranteed for 6 months when stored properly.All native purification buffers are prepared from the 5X Native PurificationBuffer and the 3 M Imidazole, as described on page 7.The Denaturing Wash Buffer pH 5.3 is prepared from the Denaturing WashBuffer (pH 6.0), as described on page 11.Resin and ColumnSpecificationsProBond™ resin is precharged with Ni2+ ions and appears blue in color. It isprovided as a 50% slurry in 20% ethanol.ProBond™ resin and purification columns have the following specifications:• Binding capacity of ProBond™ resin: 1–5 mg of protein per ml of resin• Average bead size: 45–165 microns• Pore size of purification columns: 30–35 microns• Recommended flow rate: 0.5 ml/min• Maximum flow rate: 2 ml/min• Maximum linear flow rate: 700 cm/h• Column material: Polypropylene• pH stability (long term): pH 3–13• pH stability (short term): pH 2–14ProductQualificationThe ProBond™ Purification System is qualified by purifying 2 mg of myoglobinprotein on a column and performing a Bradford assay. Protein recovery mustbe 75% or higher.vAccessory ProductsAdditionalProductsThe following products are also available for order from Invitrogen:Product QuantityCatalogNo.ProBond™ Nickel-Chelating Resin 50 ml150 mlR801-01R801-15Polypropylene columns(empty)50 R640-50Ni-NTA Agarose 10 ml25 ml R901-01 R901-15Ni-NTA Purification System 6 purifications K950-01 Ni-NTA Purification Systemwith Antibodywith Anti-Xpress™ Antibody with Anti-myc-HRP Antibody with Anti-His(C-term)-HRP Antibodywith Anti-V5-HRP Antibody 1 kit1 kit1 kit1 kitK951-01K952-01K953-01K954-01Anti-myc Antibody 50 µl R950-25 Anti-V5 Antibody 50 µl R960-25 Anti-Xpress™ Antibody 50 µl R910-25 Anti-His(C-term) Antibody 50 µl R930-25 InVision™ His-tag In-gel Stain 500 ml LC6030 InVision™ His-tag In-gelStaining Kit1 kit LC6033Pre-Cast Gels and Pre-made Buffers A large variety of pre-cast gels for SDS-PAGE and pre-made buffers for your convenience are available from Invitrogen. For details, visit our web site at or contact Technical Service (page 23).viIntroductionOverviewIntroduction The ProBond™ Purification System is designed for purification of 6xHis-tagged recombinant proteins expressed in bacteria, insect, and mammalian cells. Thesystem is designed around the high affinity and selectivity of ProBond™Nickel-Chelating Resin for recombinant fusion proteins containing six tandemhistidine residues.The ProBond™ Purification System is a complete system that includespurification buffers and resin for purifying proteins under native, denaturing,or hybrid conditions. The resulting proteins are ready for use in many targetapplications.This manual is designed to provide generic protocols that can be adapted foryour particular proteins. The optimal purification parameters will vary witheach protein being purified.ProBond™ Nickel-Chelating Resin ProBond™ Nickel-Chelating Resin is used for purification of recombinant proteins expressed in bacteria, insect, and mammalian cells from any 6xHis-tagged vector. ProBond™ Nickel-Chelating Resin exhibits high affinity and selectivity for 6xHis-tagged recombinant fusion proteins.Proteins can be purified under native, denaturing, or hybrid conditions using the ProBond™ Nickel-Chelating Resin. Proteins bound to the resin are eluted with low pH buffer or by competition with imidazole or histidine. The resulting proteins are ready for use in target applications.Binding Characteristics ProBond™ Nickel-Chelating Resin uses the chelating ligand iminodiacetic acid (IDA) in a highly cross-linked agarose matrix. IDA binds Ni2+ ions by three coordination sites.The protocols provided in this manual are generic, and may not result in 100%pure protein. These protocols should be optimized based on the bindingcharacteristics of your particular proteins.Native VersusDenaturingConditionsThe decision to purify your 6xHis-tagged fusion proteins under native ordenaturing conditions depends on the solubility of the protein and the need toretain biological activity for downstream applications.• Use native conditions if your protein is soluble (in the supernatant afterlysis) and you want to preserve protein activity.• Use denaturing conditions if the protein is insoluble (in the pellet afterlysis) or if your downstream application does not depend on proteinactivity.• Use hybrid protocol if your protein is insoluble but you want to preserveprotein activity. Using this protocol, you prepare the lysate and columnsunder denaturing conditions and then use native buffers during the washand elution steps to refold the protein. Note that this protocol may notrestore activity for all proteins. See page 14.1MethodsPreparing Cell LysatesIntroduction Instructions for preparing lysates from bacteria, insect, and mammalian cellsusing native or denaturing conditions are described below.Materials Needed You will need the following items:• Native Binding Buffer (recipe is on page 8) for preparing lysates undernative conditions• Sonicator• 10 µg/ml RNase and 5 µg/ml DNase I (optional)• Guanidinium Lysis Buffer (supplied with the system) for preparing lysatesunder denaturing conditions• 18-gauge needle• Centrifuge• Sterile, distilled water• SDS-PAGE sample buffer• Lysozyme for preparing bacterial cell lysates• Bestatin or Leupeptin, for preparing mammalian cell lysatesProcessing Higher Amount of Starting Material Instructions for preparing lysates from specific amount of starting material (bacteria, insect, and mammalian cells) and purification with 2 ml resin under native or denaturing conditions are described in this manual.If you wish to purify your protein of interest from higher amounts of starting material, you may need to optimize the lysis protocol and purification conditions (amount of resin used for binding). The optimization depends on the expected yield of your protein and amount of resin to use for purification. Perform a pilot experiment to optimize the purification conditions and then based on the pilot experiment results, scale-up accordingly.Continued on next page2Preparing Bacterial Cell Lysate—Native Conditions Follow the procedure below to prepare bacterial cell lysate under native conditions. Scale up or down as necessary.1. Harvest cells from a 50 ml culture by centrifugation (e.g., 5000 rpm for5 minutes in a Sorvall SS-34 rotor). Resuspend the cells in 8 ml NativeBinding Buffer (recipe on page 8).2. Add 8 mg lysozyme and incubate on ice for 30 minutes.3. Using a sonicator equipped with a microtip, sonicate the solution on iceusing six 10-second bursts at high intensity with a 10-second coolingperiod between each burst.Alternatively, sonicate the solution on ice using two or three 10-secondbursts at medium intensity, then flash freeze the lysate in liquid nitrogen or a methanol dry ice slurry. Quickly thaw the lysate at 37°C andperform two more rapid sonicate-freeze-thaw cycles.4. Optional: If the lysate is very viscous, add RNase A (10 µg/ml) andDNase I (5 µg/ml) and incubate on ice for 10–15 minutes. Alternatively,draw the lysate through a 18-gauge syringe needle several times.5. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.Note: Some 6xHis-tagged protein may remain insoluble in the pellet, and can be recovered by preparing a denatured lysate (page 4) followed bythe denaturing purification protocol (page 12). To recover this insolubleprotein while preserving its biological activity, you can prepare thedenatured lysate and then follow the hybrid protocol on page 14. Notethat the hybrid protocol may not restore activity in all cases, and should be tested with your particular protein.6. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or freeze at -20°C. When ready to use, proceed to theprotocol on page 7.Continued on next page3Preparing Bacterial Cell Lysate—Denaturing Conditions Follow the procedure below to prepare bacterial cell lysate under denaturing conditions:1. Equilibrate the Guanidinium Lysis Buffer, pH 7.8 (supplied with thesystem or see page 19 for recipe) to 37°C.2. Harvest cells from a 50 ml culture by centrifugation (e.g., 5000 rpm for5 minutes in a Sorvall SS-34 rotor).3. Resuspend the cell pellet in 8 ml Guanidinium Lysis Buffer from Step 1.4. Slowly rock the cells for 5–10 minutes at room temperature to ensurethorough cell lysis.5. Sonicate the cell lysate on ice with three 5-second pulses at high intensity.6. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris.Transfer the supernatant to a fresh tube.7. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or at -20°C. When ready to use, proceed to the denaturingprotocol on page 11 or hybrid protocol on page 13.Note: To perform SDS-PAGE with samples in Guanidinium Lysis Buffer, you need to dilute the samples, dialyze the samples, or perform TCAprecipitation prior to SDS-PAGE to prevent the precipitation of SDS.Harvesting Insect Cells For detailed protocols dealing with insect cell expression, consult the manual for your particular system. The following lysate protocols are for baculovirus-infected cells and are intended to be highly generic. They should be optimized for your cell lines.For baculovirus-infected insect cells, when the time point of maximal expression has been determined, large scale protein expression can be carried out. Generally, the large-scale expression is performed in 1 liter flasks seeded with cells at a density of 2 × 106 cells/ml in a total volume of 500 ml and infected with high titer viral stock at an MOI of 10 pfu/cell. At the point of maximal expression, harvest cells in 50 ml aliquots. Pellet the cells by centrifugation and store at -70°C until needed. Proceed to preparing cell lysates using native or denaturing conditions as described on the next page.Continued on next page4Preparing Insect Cell Lysate—Native Condition 1. Prepare 8 ml Native Binding Buffer (recipe on page 8) containingLeupeptin (a protease inhibitor) at a concentration of 0.5 µg/ml.2. After harvesting the cells (previous page), resuspend the cell pellet in8 ml Native Binding Buffer containing 0.5 µg/ml Leupeptin.3. Lyse the cells by two freeze-thaw cycles using a liquid nitrogen or dryice/ethanol bath and a 42°C water bath.4. Shear DNA by passing the preparation through an 18-gauge needle fourtimes.5. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris.Transfer the supernatant to a fresh tube.6. Remove 5 µl of the lysate for SDS-PAGE analysis. Store remaining lysateon ice or freeze at -20°C. When ready to use, proceed to the protocol on page 7.Preparing Insect Cell Lysate—Denaturing Condition 1. After harvesting insect cells (previous page), resuspend the cell pellet in8 ml Guanidinium Lysis Buffer (supplied with the system or see page 19for recipe).2. Pass the preparation through an 18-gauge needle four times.3. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.4. Remove 5 µl of the lysate for SDS-PAGE analysis. Store remaining lysateon ice or freeze at -20° C. When ready to use, proceed to the denaturingprotocol on page 11 or hybrid protocol on page 13.Note: To perform SDS-PAGE with samples in Guanidinium Lysis Buffer, you need to dilute the samples, dialyze the samples, or perform TCAprecipitation prior to SDS-PAGE to prevent the precipitation of SDS.Continued on next pagePreparing Mammalian Cell Lysate—Native Conditions For detailed protocols dealing with mammalian expression, consult the manual for your particular system. The following protocols are intended to be highly generic, and should be optimized for your cell lines.To produce recombinant protein, you need between 5 x 106and 1 x 107 cells. Seed cells and grow in the appropriate medium until they are 80–90% confluent. Harvest cells by trypsinization. You can freeze the cell pellet in liquid nitrogen and store at -70°C until use.1. Resuspend the cell pellet in 8 ml of Native Binding Buffer (page 8). Theaddition of protease inhibitors such as bestatin and leupeptin may benecessary depending on the cell line and expressed protein.2. Lyse the cells by two freeze-thaw cycles using a liquid nitrogen or dryice/ethanol bath and a 42°C water bath.3. Shear the DNA by passing the preparation through an 18-gauge needlefour times.4. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.5. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or freeze at -20° C. When ready to use, proceed to theprotocol on page 7.Preparing Mammalian Cell Lysates—Denaturing Conditions For detailed protocols dealing with mammalian expression, consult the manual for your particular system. The following protocols are intended to be highly generic, and should be optimized for your cell lines.To produce recombinant protein, you need between 5 x 106and 1 x 107 cells. Seed cells and grow in the appropriate medium until they are 80–90% confluent. Harvest cells by trypsinization. You can freeze the cell pellet in liquid nitrogen and store at -70°C until use.1. Resuspend the cell pellet in 8 ml Guanidinium Lysis Buffer (suppliedwith the system or see page 19 for recipe).2. Shear the DNA by passing the preparation through an 18-gauge needlefour times.3. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.4. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or freeze at -20° C until use. When ready to use, proceed to the denaturing protocol on page 11 or hybrid protocol on page 13.Note: To perform SDS-PAGE with samples in Guanidinium Lysis Buffer, you need to dilute the samples, dialyze the samples, or perform TCAprecipitation prior to SDS-PAGE to prevent the precipitation of SDS.Purification Procedure—Native ConditionsIntroduction In the following procedure, use the prepared Native Binding Buffer, NativeWash Buffer, and Native Elution Buffer, columns, and cell lysate preparedunder native conditions. Be sure to check the pH of your buffers before starting.Buffers for Native Purification All buffers for purification under native conditions are prepared from the5X Native Purification Buffer supplied with the system. Dilute and adjust the pH of the 5X Native Purification Buffer to create 1X Native Purification Buffer (page 8). From this, you can create the following buffers:• Native Binding Buffer• Native Wash Buffer• Native Elution BufferThe recipes described in this section will create sufficient buffers to perform one native purification using one kit-supplied purification column. Scale up accordingly.If you are preparing your own buffers, see page 18 for recipe.Materials Needed You will need the following items:• 5X Native Purification Buffer (supplied with the system or see page 18 forrecipe)• 3 M Imidazole (supplied with the system or see page 18 for recipe)• NaOH• HCl• Sterile distilled water• Prepared ProBond™ columns with native buffers (next page)• Lysate prepared under native conditions (page 2)Imidazole Concentration in Native Buffers Imidazole is included in the Native Wash and Elution Buffers to minimize the binding of untagged, contaminating proteins and increase the purity of the target protein with fewer wash steps. Note that, if your level of contaminating proteins is high, you may add imidazole to the Native Binding Buffer.If your protein does not bind well under these conditions, you can experiment with lowering or eliminating the imidazole in the buffers and increasing the number of wash and elution steps.Continued on next page1X Native Purification Buffer To prepare 100 ml 1X Native Purification Buffer, combine:• 80 ml of sterile distilled water• 20 ml of 5X Native Purification Buffer (supplied with the system or see page 18 for recipe)Mix well and adjust pH to 8.0 with NaOH or HCl.Native Binding Buffer Without ImidazoleUse 30 ml of the 1X Native Purification Buffer (see above for recipe) for use as the Native Binding Buffer (used for column preparation, cell lysis, and binding).With Imidazole (Optional):You can prepare the Native Binding Buffer with imidazole to reduce the binding of contaminating proteins. (Note that some His-tagged proteins may not bind under these conditions.).To prepare 30 ml Native Binding Buffer with 10 mM imidazole, combine: • 30 ml of 1X Native Purification Buffer• 100 µl of 3 M Imidazole, pH 6.0Mix well and adjust pH to 8.0 with NaOH or HCl.Native Wash Buffer To prepare 50 ml Native Wash Buffer with 20 mM imidazole, combine:• 50 ml of 1X Native Purification Buffer• 335 µl of 3 M Imidazole, pH 6.0Mix well and adjust pH to 8.0 with NaOH or HCl.Native Elution Buffer To prepare 15 ml Native Elution Buffer with 250 mM imidazole, combine:• 13.75 ml of 1X Native Purification Buffer• 1.25 ml of 3 M Imidazole, pH 6.0Mix well and adjust pH to 8.0 with NaOH or HCl.Continued on next pageDo not use strong reducing agents such as DTT with ProBond™ columns. DTTreduces the nickel ions in the resin. In addition, do not use strong chelatingagents such as EDTA or EGTA in the loading buffers or wash buffers, as thesewill strip the nickel from the columns.Be sure to check the pH of your buffers before starting.PreparingProBond™ ColumnWhen preparing a column as described below, make sure that the snap-off capat the bottom of the column remains intact. To prepare a column:1. Resuspend the ProBond™ resin in its bottle by inverting and gentlytapping the bottle repeatedly.2. Pipet or pour 2 ml of the resin into a 10-ml Purification Columnsupplied with the kit. Allow the resin to settle completely by gravity(5-10 minutes) or gently pellet it by low-speed centrifugation (1 minuteat 800 ×g). Gently aspirate the supernatant.3. Add 6 ml of sterile, distilled water and resuspend the resin byalternately inverting and gently tapping the column.4. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant.5. For purification under Native Conditions, add 6 ml Native BindingBuffer (recipe on page 8).6. Resuspend the resin by alternately inverting and gently tapping thecolumn.7. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant.8. Repeat Steps 5 through 7.Storing PreparedColumnsTo store a column containing resin, add 0.02% azide or 20% ethanol as apreservative and cap or parafilm the column. Store at room temperature.Continued on next pagePurification Under Native Conditions Using the native buffers, columns and cell lysate, follow the procedure below to purify proteins under native conditions:1. Add 8 ml of lysate prepared under native conditions to a preparedPurification Column (page 9).2. Bind for 30–60 minutes using gentle agitation to keep the resinsuspended in the lysate solution.3. Settle the resin by gravity or low speed centrifugation (800 ×g), andcarefully aspirate the supernatant. Save supernatant at 4°C forSDS-PAGE analysis.4. Wash with 8 ml Native Wash Buffer (page 8). Settle the resin by gravityor low speed centrifugation (800 ×g), and carefully aspirate thesupernatant. Save supernatant at 4°C for SDS-PAGE analysis.5. Repeat Step 4 three more times.6. Clamp the column in a vertical position and snap off the cap on thelower end. Elute the protein with 8–12 ml Native Elution Buffer (seepage 2). Collect 1 ml fractions and analyze with SDS-PAGE.Note: Store the eluted fractions at 4°C. If -20°C storage is required, addglycerol to the fractions. For long term storage, add protease inhibitors to the fractions.If you wish to reuse the resin to purify the same recombinant protein, wash the resin with 0.5 M NaOH for 30 minutes and equilibrate the resin in a suitable binding buffer. If you need to recharge the resin, see page 17.Purification Procedure—Denaturing ConditionsIntroduction Instructions to perform purification using denaturing conditions with prepareddenaturing buffers, columns, and cell lysate are described below.Materials Needed You will need the following items:• Denaturing Binding Buffer (supplied with the system or see page 19 forrecipe)• Denaturing Wash Buffer, pH 6.0 (supplied with the system or see page 19 forrecipe) and Denaturing Wash Buffer, pH 5.3 (see recipe below)• Denaturing Elution Buffer (supplied with the system or see page 20 forrecipe)• Prepared ProBond™ columns with Denaturing buffers (see below)• Lysate prepared under denaturing conditions (page 11)Preparing the Denaturing Wash Buffer pH 5.3 Using a 10 ml aliquot of the kit-supplied Denaturing Wash Buffer (pH 6.0), mix well, and adjust the pH to 5.3 using HCl. Use this for the Denaturing Wash Buffer pH 5.3 in Step 5 next page.Be sure to check the pH of your buffers before starting. Note that thedenaturing buffers containing urea will become more basic over time. PreparingProBond™ ColumnWhen preparing a column as described below, make sure that the snap-off capat the bottom of the column remains intact.If you are reusing the ProBond™ resin, see page 17 for recharging protocol.To prepare a column:1. Resuspend the ProBond™ resin in its bottle by inverting and gentlytapping the bottle repeatedly.2. Pipet or pour 2 ml of the resin into a 10-ml Purification Columnsupplied with the kit. Allow the resin to settle completely by gravity(5-10 minutes) or gently pellet it by low-speed centrifugation (1 minuteat 800 ×g). Gently aspirate the supernatant.3. Add 6 ml of sterile, distilled water and resuspend the resin byalternately inverting and gently tapping the column.4. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant.5. For purification under Denaturing Conditions, add 6 ml of DenaturingBinding Buffer.6. Resuspend the resin by alternately inverting and gently tapping thecolumn.7. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant. Repeat Steps 5 through 7.Continued on next pagePurification Procedure—Denaturing Conditions, ContinuedPurification Under Denaturing Conditions Using the denaturing buffers, columns, and cell lysate, follow the procedure below to purify proteins under denaturing conditions:1. Add 8 ml lysate prepared under denaturing conditions to a preparedPurification Column (page 11).2. Bind for 15–30 minutes at room temperature using gentle agitation (e.g.,using a rotating wheel) to keep the resin suspended in the lysatesolution. Settle the resin by gravity or low speed centrifugation (800 ×g), and carefully aspirate the supernatant.3. Wash the column with 4 ml Denaturing Binding Buffer supplied with thekit by resuspending the resin and rocking for two minutes. Settle theresin by gravity or low speed centrifugation (800 ×g), and carefullyaspirate the supernatant. Save supernatant at 4°C for SDS-PAGEanalysis. Repeat this step one more time.4. Wash the column with 4 ml Denaturing Wash Buffer, pH 6.0 supplied inthe kit by resuspending the resin and rocking for two minutes. Settle the resin by gravity or low speed centrifugation (800 ×g), and carefullyaspirate the supernatant. Save supernatant at 4°C for SDS-PAGEanalysis. Repeat this step one more time.5. Wash the column with 4 ml Denaturing Wash Buffer pH 5.3 (see recipeon previous page) by resuspending the resin and rocking for 2 minutes.Settle the resin by gravity or low speed centrifugation (800 ×g), andcarefully aspirate the supernatant. Save supernatant at 4°C for SDS-PAGE analysis. Repeat this step once more for a total of two washes with Denaturing Wash Buffer pH 5.3.6. Clamp the column in a vertical position and snap off the cap on thelower end. Elute the protein by adding 5 ml Denaturing Elution Buffersupplied with the kit. Collect 1 ml fractions and monitor the elution bytaking OD280readings of the fractions. Pool the fractions that contain the peak absorbance and dialyze against 10 mM Tris, pH 8.0, 0.1% Triton X-100 overnight at 4°C to remove the urea. Concentrate the dialyzedmaterial by any standard method (i.e., using 10,000 MW cut-off, low-protein binding centrifugal instruments or vacuum concentrationinstruments).If you wish to reuse the resin to purify the same recombinant protein, wash the resin with 0.5 M NaOH for 30 minutes and equilibrate the resin in a suitable binding buffer. If you need to recharge the resin, see page 17.。




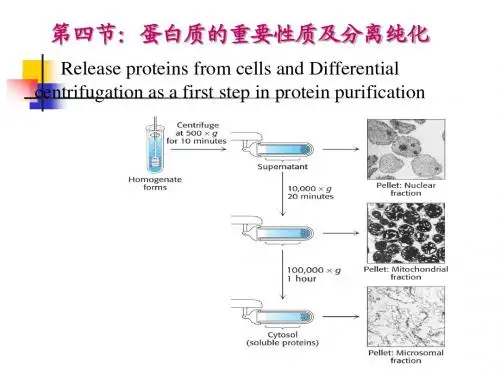

Protein PurificationHandbook18-1132-29Edition ABHiTrap, Sepharose, STREAMLINE, Sephadex, MonoBeads, Mono Q,Mono S, MiniBeads, RESOURCE, SOURCE, Superdex, Superose, HisTrap, HiLoad, HiPrep, INdEX, BPG, BioProcess, FineLINE, MabTrap, MAbAssistant, Multiphor, FPLC, PhastSystem and ÄKTA are trademarks of Amersham Pharmacia Biotech Limitedor its subsidiaries.Amersham is a trademark of Nycomed Amersham plcPharmacia and Drop Design are trademarks of Pharmacia & Upjohn Inc Coomassie is a trademark of ICI plcAll goods and services are sold subject to the terms and conditions of sale of the company within the Amersham Pharmacia Biotech group which supplies them. A copy of these terms and conditions of sale is available on request.© Amersham Pharmacia Biotech AB 1999-All rights reserved.Amersham Pharmacia Biotech ABSE-751 84 Uppsala SwedenAmersham Pharmacia Biotech UK Limited Amersham Place Little Chalfont Buckinghamshire England HP7 9NA Amersham Pharmacia Biotech Inc800 Centennial Avenue PO Box 1327 Piscataway NJ 08855 USAProtein Purification HandbookContents Introduction (7)Chapter 1Purification Strategies - A Simple Approach (9)Preparation (10)Three Phase Purification Strategy (10)General Guidelines for Protein Purification (12)Chapter 2 Preparation (13)Before You Start (13)Sample Extraction and Clarification (16)Chapter 3Three Phase Purification Strategy (19)Principles (19)Selection and Combination of Purification Techniques (20)Sample Conditioning (26)Chapter 4Capture (29)Chapter 5Intermediate Purification (37)Chapter 6Polishing (40)Chapter 7Examples of Protein Purification Strategies (45)Three step purification of a recombinant enzyme (45)Three step purification of a recombinant antigen binding fragment (49)Two step purification of a monoclonal antibody (54)One step purification of an integral membrane protein (57)Chapter 8Storage Conditions (61)Extraction and Clarification Procedures (62)Chapter 9Principles and Standard Conditions for Purification Techniques (73)Ion exchange (IEX) (73)Hydrophobic interaction (HIC) (79)Affinity (AC) (85)Gel filtration (GF) (88)Reversed phase (RPC) (92)Expanded bed adsorption (EBA) (95)IntroductionThe development of techniques and methods for protein purification has been an essential pre-requisite for many of the advancements made in biotechnology. This booklet provides advice and examples for a smooth path to protein purification. Protein purification varies from simple one-step precipitation procedures to large scale validated production processes. Often more than one purification step is necessary to reach the desired purity. The key to successful and efficient protein purification is to select the most appropriate techniques, optimise their performance to suit the requirements and combine them in a logical way to maximise yield and minimise the number of steps required.Most purification schemes involve some form of chromatography. As a result chromatography has become an essential tool in every laboratory where protein purification is needed. The availability of different chromatography techniques with different selectivities provides a powerful combination for the purification of any biomolecule.Recombinant DNA developments over the past decade have revolutionised the production of proteins in large quantities. Proteins can even be produced in forms which facilitate their subsequent chromatographic purification. However, this has not removed all challenges. Host contaminants are still present and problems related to solubility, structural integrity and biological activity can still exist. Although there may appear to be a great number of parameters to consider, with a few simple guidelines and application of the Three Phase Purification Strategy the process can be planned and performed simply and easily, with only a basic knowledge of the details of chromatography techniques.78Chapter 1Purification Strategies- a simple approachApply a systematic approach to development of a purification strategy. The first step is to describe the basic scenario for the purification. General considerations answer questions such as: What is the intended use of the product? What kind of starting material is available and how should it be handled? What are the purity issues in relation to the source material and intended use of the final product? What has to be removed? What must be removed completely? What will be the final scale of purification? If there is a need for scale-up, what consequences will this have on the chosen purification techniques? What are the economical constraints and what resources and equipment are available?Most purification protocols require more than one step to achieve the desired level of product purity. This includes any conditioning steps necessary to transfer the product from one technique into conditions suitable to perform the next technique. Each step in the process will cause some loss of product. For example, if a yield of 80% in each step is assumed, this will be reduced to only 20% overall yield after 8 processing steps as shown in Figure 1. Consequently, to reach the targets for yield and purity with the minimum number of steps and the simplest possible design, it is not efficient to add one step to another until purity requirements have been fulfilled. Occasionally when a sample is readily available purity can be achieved by simply adding or repeating steps. However, experience shows that, even for the most challenging applications, high purity and yield can be achieved efficiently in fewer than four well-chosen and optimised purification steps. Techniques should be organised in a logical sequence to avoid the need for conditioning steps and the chromatographic techniques selected appropriately to use as few purification steps as possible.Limit the number of steps in a purification procedure910Fig.1.Yields from multi-step purifications.PreparationThe need to obtain a protein, efficiently, economically and in sufficient purity and quantity, applies to every purification. It is important to set objectives for purity,quantity and maintenance of biological activity and to define the economical and time framework for the work. All information concerning properties of the target protein and contaminants will help during purification development. Some simple experiments to characterise the sample and target molecule are an excellent investment. Development of fast and reliable analytical assays is essential to follow the progress of the purification and assess its effectiveness. Sample preparation and extraction procedures should be developed prior to the first chromatographic purification step.With background information, assays and sample preparation procedures in place the Three Phase Purification Strategy can be considered.Three Phase Purification Strategy Imagine the purification has three phases Capture, IntermediatePurification and Polishing.In the Three Phase Strategy specific objectives are assigned to each step within the process:In the capture phase the objectives are to isolate, concentrate and stabilise the target product.During the intermediate purification phase the objective is to remove most of the bulk impurities such as other proteins and nucleic acids, endotoxins and viruses.In the polishing phase the objective is to achieve high purity by removing any remaining trace impurities or closely related substances.The selection and optimum combination of purification techniques for Capture,Intermediate Purification and Polishing is crucial to ensure fast method development, a shorter time to pure product and good economy.108060402012345678Number of steps 95% / step90% / step 85% / step 80% / step 75% / stepYield (%)The final purification process should ideally consist of sample preparation, including extraction and clarification when required, followed by three major purification steps, as shown in Figure 2. The number of steps used will always depend upon the purity requirements and intended use for the protein.Fig. 2.Preparation and the Three Phase Purification Strategy11Guidelines for Protein PurificationThe guidelines for protein purification shown here can be applied to any purification process and are a suggestion as to how a systematic approach can be applied to the development of an effective purification strategy. As a reminder these guidelines will be highlighted where appropriate throughout the following chapters.Define objectivesfor purity, activity and quantity required of final product to avoid over or under developing a methodDefine properties of target protein and critical impuritiesto simplify technique selection and optimisationDevelop analytical assaysfor fast detection of protein activity/recovery and to work efficientlyMinimise sample handling at every stageto avoid lengthy procedures which risk losing activity/reducing recovery Minimise use of additivesadditives may need to be removed in an extra purification step or may interfere with activity assaysRemove damaging contaminants earlyfor example, proteasesUse a different technique at each stepto take advantage of sample characteristics which can be used for separation (size, charge, hydrophobicity, ligand specificity)Minimise number of stepsextra steps reduce yield and increase time, combine steps logicallyKEEP IT SIMPLE!12Chapter 2PreparationBefore You StartThe need to obtain a protein, efficiently, economically and in sufficient purity and quantity, applies to any purification, from preparation of an enriched protein extract for biochemical characterisation to large scale production of a therapeutic recombinant protein. It is important to set objectives for purity and quantity, maintenance of biological activity and economy in terms of money and time. Purity requirements must take into consideration the nature of the source material, the intended use of the final product and any special safety issues. For example, it is important to differentiate between contaminants which must be removed and those which can be tolerated. Other factors can also influence the prioritisation of objectives. High yields are usually a key objective, but may be less crucial in cases where a sample is readily available or product is required only in small quantities. Extensive method development may be impossible without resources such as an ÄKTA™design chromatography system. Similarly, time pressure combined with a slow assay turnaround will steer towards less extensive scouting and optimisation. All information concerning properties of the target protein and contaminants will help during purification development, allowing faster and easier technique selection and optimisation, and avoiding conditions which may inactivate the target protein.Development of fast and reliable analytical assays is essential to follow the progress of the purification and assess effectiveness (yield, biological activity, recovery).Define objectivesGoal:To set minimum objectives for purity and quantity, maintenance of biological activity and economy in terms of money and time.Define purity requirements according to the final use of the product. Purity requirement examples are shown below.Extremely high > 99%Therapeutic use, in vivo studiesHigh 95- 99 %X-ray crystallography and most physico-chemicalcharacterisation methodsModerate < 95 %Antigen for antibody productionN-terminal sequencing13Identify 'key' contaminantsIdentify the nature of possible remaining contaminants as soon aspossible.The statement that a protein is >95% pure (i.e. target protein constitutes 95% of total protein) is far from a guarantee that the purity is sufficient for an intended application. The same is true for the common statement "the protein was homogenous by Coomassie™ stained SDS-PAGE". Purity of 95% may be acceptable if the remaining 5% consists of harmless impurities. However, even minor impurities which may be biologically active could cause significant problems in both research and therapeutic applications. It is therefore important to differentiate between contaminants which must be removed completely and those which can be reduced to acceptable levels. Since different types of starting material will contain different contaminant profiles they will present different contamination problems.It is better to over-purify than to under-purify.Although the number of purification steps should be minimised, thequality of the end product should not be compromised. Subsequent results might be questioned if sample purity is low and contaminants are unknown.Contaminants which degrade or inactivate the protein or interfere withanalyses should be removed as early as possible.The need to maintain biological activity must be considered at every stage during purification development. It is especially beneficial if proteases are removed and target protein transferred into a friendly environment during the first step.Economy is a very complex issue. In commercial production the time to market can override issues such as optimisation for recovery, capacity or speed. Robustness and reliability are also of great concern since a batch failure can have major consequences.It may be necessary to use analytical techniques targetted towards specific conta-minants in order to demonstrate that they have been removed to acceptable levels. 14Define properties of target protein and critical impurities Goal:To determine a 'stability window' for the target protein for easier selection and optimisation of techniques and to avoid protein inactivation during purification.Check target protein stability window for at least pH and ionic strength. All information concerning the target protein and contaminant properties will help to guide the choice of separation techniques and experimental conditions for purification. Database information for the target, or related proteins, may give size, isoelectric point (pI) and hydrophobicity or solubility data. Native one and two dimensional PAGE can indicate sample complexity and the properties of the target protein and major contaminants. Particularly important is a knowledge of the stability window of the protein so that irreversible inactivation is avoided. Itis advisable to check the target protein stability window for at least pH and ionic strength. Table 1 shows how different target protein properties can affect a purification strategy.Table 1.Protein properties and their effect on development of purification strategies. Sample and target protein properties Influence on purification strategyTemperature stability Need to work rapidly at lowered temperaturepH stability Selection of buffers for extraction and purificationSelection of conditions for ion exchange, affinity orreversed phase chromatographyOrganic solvents stability Selection of conditions for reversed phasechromatographyDetergent requirement Consider effects on chromatographic steps and the needfor detergent removal. Consider choice of detergent.Salt (ionic strength)Selection of conditions for precipitation techniques andhydrophobic interaction chromatographyCo-factors for stability or activity Selection of additives, pH, salts, buffersProtease sensitivity Need for fast removal of proteases or addition ofinhibitorsSensitivity to metal ions Need to add EDTA or EGTA in buffersRedox sensitivity Need to add reducing agentsMolecular weight Selection of gel filtration mediaCharge properties Selection of ion exchange conditionsBiospecific affinity Selection of ligand for affinity mediumPost translational modifications Selection of group specific affinity medium Hydrophobicity Selection of medium for hydrophobic interactionchromatography15Develop analytical assaysGoal:To follow the progress of a purification, to assess effectiveness (yield, biological activity, recovery) and to help during optimisation.Select assays which are fast and reliable.To progress efficiently during method development the effectiveness of each step should be assessed. The laboratory should have access to the following assays:• A rapid, reliable assay for the target protein• Purity determination• Total protein determination• Assays for impurities which must be removedThe importance of a reliable assay for the target protein cannot be over- emphasised. When testing chromatographic fractions ensure that the buffers used for separation do not interfere with the assay. Purity of the target protein is most often estimated by SDS-PAGE, capillary electrophoresis, reversed phase chromatography or mass spectrometry. Lowry or Bradford assays are used most frequently to determine the total protein.The Bradford assay is particularly suited to samples where there is a high lipid content which may interfere with the Lowry assay.For large scale protein purification the need to assay for target proteins and critical impurities is often essential. In practice, when a protein is purified for research purposes, it is too time consuming to identify and set up specific assays for harmful contaminants. A practical approach is to purify the protein to a certain level, and then perform SDS-PAGE after a storage period to check for protease cleavage. Suitable control experiments, included within assays forbio-activity, will help to indicate if impurities are interfering with results.Sample Extraction and Clarification Minimise sample handlingMinimise use of additivesRemove damaging contaminants earlyDefinition:Primary isolation of target protein from source material.Goal:Preparation of a clarified sample for further purification. Removal of particulate matter or other contaminants which are not compatible with chromatography.16The need for sample preparation prior to the first chromatographic step is dependent upon sample type. In some situations samples may be taken directly to the first capture step. For example cell culture supernatant can be applied directly to a suitable chromatographic matrix such as Sepharose™ Fast Flow and may require only a minor adjustment of the pH or ionic strength. However, it is most often essential to perform some form of sample extraction and clarification procedure.If sample extraction is required the chosen technique must be robust and suitable for all scales of purification likely to be used. It should be noted that a technique such as ammonium sulphate precipitation, commonly used in small scale, may be unsuitable for very large scale preparation. Choice of buffers and additives must be carefully considered if a purification is to be scaled up. In these cases inexpensive buffers, such as acetate or citrate, are preferable to the more complex compositions used in the laboratory. It should also be noted that dialysis and other common methods used for adjustment of sample conditions are unsuitable for very large or very small samples.For repeated purification, use an extraction and clarification techniquethat is robust and able to handle sample variability. This ensures areproducible product for the next purification step despite variability instarting material.Use additives only if essential for stabilisation of product or improvedextraction. Select those which are easily removed. Additives may need tobe removed in an extra purification step.Use pre-packed columns of Sephadex™ G-25 gel filtration media, forrapid sample clean-up at laboratory scale, as shown in Table 2.Table 2.Pre-packed columns for sample clean-up.Pre-packed column Sample volume Sample volume Code No.loading per run recovery per runHiPrep™Desalting 26/10 2.5 -15 ml7.5 - 20 ml17-5087-01HiTrap Desalting0.25 - 1.5 ml 1.0 - 2.0 ml17-1408-01Fast Desalting PC 3.2/100.05 - 0.2 ml0.2 - 0.3 ml17-0774-01PD-10 Desalting 1.5 - 2.5 ml 2.5 - 3.5 ml17-0851-01 Sephadex G-25 gel filtration media are used at laboratory and production scale for sample preparation and clarification of proteins >5000. Sample volumes of up to 30%, or in some cases, 40% of the total column volume are loaded. In a single step, the sample is desalted, exchanged into a new buffer, and low molecular weight materials are removed. The high volume capacity, relative insensitivity to sample concentration, and speed of this step enable very large sample volumes to be processed rapidly and efficiently. Using a high sample volume load results in a separation with minimal sample dilution (approximately 1:1.4). Chapter 8 contains further details on sample storage, extraction and clarification procedures.17Sephadex G-25 is also used for sample conditioning i.e. rapid adjustment of pH, buffer exchange and desalting between purification steps.Sephadex G-25 gel filtrationFor fast group separations between high and low molecular weight substances Typical flow velocity 60 cm/h (Sephadex G-25 SuperFine, Sephadex G-25 Fine), 150 cm/h (Sephadex G-25 Medium).If large sample volumes will be handled or the method scaled-up in the future, consider using STREAMLINE™ expanded bed adsorption. This technique is particularly suited for large scale recombinant protein and monoclonal antibody purification. The crude sample containing particles can be applied to the expanded bed without filtration or centrifugation. STREAMLINE adsorbents are specially designed for use in STREAMLINE columns. Together they enable the high flow rates needed for high productivity in industrial applications of fluidised beds. The technique requires no sample clean up and so combines sample preparation and capture in a single step. Crude sample is applied to an expanded bed STREAMLINE media. Target proteins are captured whilst cell debris, cells, particulate matter, whole cells, and contaminants pass through. Flow is reversed and the target proteins are desorbed in the elution buffer.STREAMLINE (IEX, AC, HIC)For sample clean-up and capture direct from crude sample.STREAMLINE adsorbents are designed to handle feed directly from both fermentation homogenate and crude feedstock from cell culture/fermentation at flow velocities of 200 - 500 cm/h, according to type and application.Particle size: 200 µmNote:cm/h: flow velocity (linear flow rate) = volumetric flow rate/cross sectional area of column.18Chapter 3Three Phase Purification StrategyPrinciplesWith background information, assays, and sample preparation and extraction procedures in place the Three Phase Purification Strategy can be applied (Figure 3). This strategy is used as an aid to the development of purification processes for therapeutic proteins in the pharmaceutical industry and is equally efficient as an aid when developing purification schemes in the research laboratory.Fig. 3.Preparation and the Three Phase Purification Strategy.Assign a specific objective to each step within the purification process.In the Three Phase Strategy a specific objective is assigned to each step. The purification problem associated with a particular step will depend greatly upon the properties of the starting material. Thus, the objective of a purification step will vary according to its position in the process i.e. at the beginning for isolation of product from crude sample, in the middle for further purification of partially purified sample, or at the end for final clean up of an almost pure product.The Three Phase Strategy ensures faster method development, a shorter time to pure product and good economy.In the capture phase the objectives are to isolate, concentrate and stabilise the target product. The product should be concentrated and transferred to an environment which will conserve potency/activity. At best, significant removal of other critical contaminants can also be achieved.19During the intermediate purification phase the objectives are to remove most of the bulk impurities,such as other proteins and nucleic acids, endotoxins and viruses.In the polishing phase most impurities have already been removed except for trace amounts or closely related substances. The objective is to achieve final purity.It should be noted that this Three Phase Strategy does not mean that all strategies must have three purification steps. For example, capture and intermediate purification may be achievable in a single step, as may intermediate purification and polishing. Similarly, purity demands may be so low that a rapid capture step is sufficient to achieve the desired result, or the purity of the starting material may be so high that only a polishing step is needed. For purification of therapeutic proteins a fourth or fifth purification step may be required to fulfil the highest purity and safety demands.The optimum selection and combination of purification techniques for Capture, Intermediate Purification and Polishing is crucial for an efficient purification process.Selection and Combination ofPurification TechniquesMinimise sample handlingMinimise number of stepsUse different techniques at each stepGoal:Fastest route to a product of required purity.For any chromatographic separation each different technique will offer different performance with respect to recovery, resolution, speed and capacity. A technique can be optimised to focus on one of these parameters, for example resolution, or to achieve the best balance between two parameters, such as speed and capacity.A separation optimised for one of these parameters will produce results quite different in appearance from those produced using the same technique, but focussed on an alternative parameter. See, for example, the results shown on page 49 where ion exchange is used for a capture and for a polishing step.20Select a technique to meet the objectives for the purification step. Capacity,in the simple model shown, refers to the amount of target protein loaded during purification. In some cases the amount of sample which can be loaded may be limited by volume (as in gel filtration) or by large amounts of contaminants rather than the amount of the target protein.Speed is of the highest importance at the beginning of a purification where contaminants such as proteases must be removed as quickly as possible. Recovery becomes increasingly important as the purification proceeds because of the increased value of the purified product. Recovery is influenced by destructive processes in the sample and unfavourable conditions on the column. Resolution is achieved by the selectivity of the technique and the efficiency of the chromatographic matrix to produce narrow peaks. In general, resolution is most difficult to achieve in the final stages of purification when impurities and target protein are likely to have very similar properties.Every technique offers a balance between resolution, speed, capacity and recovery and should be selected to meet the objectives for each purification step. In general, optimisation of any one of these four parameters can only be achieved at the expense of the others and a purification step will be a compromise. The importance of each parameter will vary depending on whether a purification step is used for capture, intermediate purification or polishing. This will steer the optimisation of the critical parameters, as well as the selection of the most suitable media for the step.Proteins are purified using chromatographic purification techniques which separate according to differences in specific properties, as shown in Table 3. Table 3.Protein properties used during purification.Protein property TechniqueCharge Ion exchange (IEX)Size Gel filtration (GF)Hydrophobicity Hydrophobic interaction (HIC),reversed phase (RPC)Biorecognition (ligand specificity)Affinity (AC)Charge, ligand specificity or hydrophobicity Expanded bed adsorption (EBA) follows theprinciples of AC, IEX or HIC21。


组织工程医疗产品第6部分:部分:II 型胶原蛋白1范围本标准规定了用于外科植入物和组织工程医疗产品I 型胶原蛋白的要求、试验方法、检验规则、标志、使用说明书、包装、运输和贮存等。
本标准适用于I 型胶原蛋白。
I 型胶原蛋白可以用于制备外科植入物和组织工程医疗产品。
2规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。
凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。
凡是不注日期的引用文件,其最新版本适用于本标准。
GB191-2000包装储运图示标志GB/T 5009.6-2003食品中脂肪的测定方法GB 9969.1-1998工业产品使用说明书总则GB/T 16886.1-2001医疗器械生物学评价第1部分:评价与试验GB/T 16886.3-1997医疗器械生物学评价第3部分:遗传毒性、致癌性和生殖毒性试验GB/T 16886.4-2003医疗器械生物学评价第4部分:与血液相互作用试验选择GB/T 16886.5-2003医疗器械生物学评价第5部分:细胞毒性试验;体外法GB/T 16886.6-1997医疗器械生物学评价第6部分:植入后局部反应试验GB/T 16886.10-2000医疗器械生物学评价第10部分:刺激与致敏性试验GB/T 16886.11-1997医疗器械生物学评价第11部分:全身毒性试验GB/T 16886.12-2000医疗器械生物学评价第12部分:样品制备与参照样品中华人民共和国药典(2005年版)YY/T 0313-1998医用高分子制品包装、标志、运输和贮存YY 0466-2003医疗器械用于医疗器械标签、标记和提供信息的符号3术语本标准采用下列术语3.1I 型胶原蛋白(型胶原蛋白(type type I collagen collagen))I 型胶原蛋白是在动物体中发现的一种结构蛋白,是胶原纤维组分中的一部分。

蛋白质相互作用的方法
蛋白质相互作用的方法可以分为以下几种:
1. 体外共沉淀法(Co-Immunoprecipitation):利用抗体与目标蛋白质结合后,通过共沉淀的方式来寻找与目标蛋白质相互作用的其他蛋白质。
2. 酵母双杂交法(Yeast Two-Hybrid):利用酵母细胞中的转录激活子域和靶蛋白质的相互作用来筛选相互作用蛋白。
3. 蛋白质亲和纯化法(Protein Affinity Purification):构建蛋白质相互作用的亲和纯化系统,将目标蛋白质与其他潜在相互作用蛋白质结合,并通过亲和柱等手段分离纯化相互作用蛋白质。
4. 融合蛋白技术(Fusion Protein):利用蛋白质融合技术,将目标蛋白质与报告标记或纯化标签蛋白质进行融合,通过检测标签蛋白质来确定相互作用的蛋白质。
5. 走向复合物的质谱法(Mass Spectrometry):将目标蛋白质与其他潜在相互作用蛋白质共同提取纯化,然后通过质谱分析来鉴定复合物中的蛋白质。
这些方法可以单独或组合使用来研究蛋白质的相互作用。
包涵体表达蛋白的纯化方法Purification of Expressed Proteins from Inclusion Bodies试剂和溶液Cell lysis buffer I : 50 mM Tris-Cl (pH 8.0), 1 mM EDTA (pH 8.0), 100 mM NaCl;Cell lysis buffer II: 50 mM Tris-Cl (pH 8.0), 10 mM EDTA (pH 8.0), 100 mM NaCl, 0.5% (v/v) Triton X-100; 脱氧胆酸(蛋白纯); HCl (12 M) (浓盐酸);包含体溶解缓冲液I (用前准备) :50 mM Tris-Cl (pH 8.0), 1 mM EDTA (pH 8.0), 100 mM NaCl, 8 M urea, 0.1 M PMSF or Pefabloc SC;包含体溶解缓冲液II: 50 mM KH2PO4 (pH 10.7), 1 mM EDTA (pH 8.0), 50 mM NaCl; KOH (10 N); PMSF (phenylmethylsulfonyl fluoride苯甲基磺酰氟)(17.4 mg/ml溶于异丙醇中-20°C)注:可选择PMSF或Pefabloc SC(4-2-胺乙基苯磺酰氟盐酸盐),在缓冲溶液中无毒稳定.Tris-Cl (0.1 M, pH 8.5)尿素用于第7步方法2.准备0.1 M Tris-Cl (pH 8.5)和浓度递增的尿素(e.g., 0.5, 1, 2, and 5 M). 因为尿素在水溶液中分解,用新配的尿素立即使用。
载体和寄主:表达包含体形式的目的蛋白的大肠杆菌(1L培养物)。
酶和缓冲液:DNase I (1 mg/ml in 20 mM Tris-Cl [pH 7.8]), Lysozyme(溶菌酶)Gels/Loading Buffers: 10%含SDS聚丙烯酰胺凝胶, 1×SDS 凝胶上样缓冲液:50 mM Tris-Cl (pH 6.8), 2% (w/v) SDS分析纯, 0.1% (w/v) 溴酚蓝;在第13步用缓冲液前,加1 M二硫苏糖醇(dithiothreitol, DTT)储液到终浓度100 mM.2x SDS上样缓冲液: 100 mM Tris-Cl (pH 6.8), 4% (w/v) SDS分析纯, 0.2% (w/v) 溴苯酚兰20% 甘油;在第7和14步用前加1 M二硫苏糖醇(dithiothreitol, DTT)储液到终浓度100 mM.离心机/转子/离心管:Sorvall SLC-1500 rotor (4°C);其他材料:玻璃棒(磨光的), pH试纸.实验步骤:1.用预称重的离心管于4°C离心1L表达目的蛋白的E. coli培养液5000g(5500rpm in a Sorvall SLC-1500 rotor) 15 min.重要:完成步骤2-4 在4°C.2.倒掉上清确定E. coli沉淀重量. 每克(wet weight) E. coli加3 ml cell lysis buffer I温和蜗旋或通过磨光的玻璃棒搅动来重悬沉淀.3.每克E. coli加4 µl 100 mM PMSF 或Pefabloc SC及80µl的10g/l lysozyme.搅动混匀20min.4.继续搅拌, 每克E. coli加4 mg脱氧胆酸.5.置于37°C悬浮,偶尔用玻璃棒搅动.当溶解产物变粘稠,每克E. coli加20µl 1mg/ml DNase I.6.室温静置溶菌产物直到不再粘稠(约30 minutes).7.纯化和洗涤包含体有下面两种方法.Method 1:用Triton X-100 复性包含体a.高速离心细胞溶解产物4°C 15 min.b.倒掉上清. 在4°C重悬沉淀于9倍体积cell lysis buffer II.c.室温静置悬浮液5min。
蛋白质纯化的原理和方法(Protein Purification Principles and Methods)Proteins•Complex, polymeric, asymmetric and sensitive molecules•Contain covalent bound prosthetic groups and non-covalent bound cofactors •Many non-covalent bounds e.g. Hydrogen-Bounds, Dipol-Interactions and Hydrophobic-Interactions•“Weak” interactions are important for structure and function (activity) of the protein In most cases the purification must be gentle!Before the purification…•Cultivation of bacteria•Cell disruption: Periplasmic and cytoplasmic proteins are released •Centrifugation leads to a soluble fraction(supernatant) which contains all soluble periplasmic and cytoplasmic proteins and a membrane fraction from which membrane bound proteins can be solubilised with detergents (e.g. Triton X-100)•The soluble or membrane fraction are the start point of the further purification by chromatographyCell disruption:French PressLysozymeUltrasonicFrench PressMembrane Proteins•Peripheral membrane proteins: in most cases soluble in buffers with high or low ionic strength or high pH•Integral membrane proteins: they contain trans membrane helices and must be solubilised to conserve conformation and function of the proteinSolubilisationof integral membrane proteins•Solubilisationof proteins is done with a detergents concentration above the CMC to ensure the incorporation of membrane lipid into detergent micelles.•CMC = critical micelle concentrationdepends on temperature, ionic strength and pH of the buffer and concentration of uncharged substances like urea or alcoholSome detergents•Ionic detergents:Sodium-Dodecylsulfate:denatures Proteins (SDS-PAGE)Na-Deoxycholate: preticipatesby pH<6.8preticipatewith Ca2+or Mg2+In General: No ionic Detergents in purifications with depend on the charge of the proteins. •Non-ionic detergents:Triton X-100 Phenyl groups strong Absorbance at 280 nmTween 20 like Triton X-100 not dialyzable•Zwitter-ionic detergentsChaps dialyzableWhich proteins are purified?•Metabolic pathways•Energy productionAim: biochemical characterisation (Reactivity, subunit composition, organic and inorganic cofactors, 3D structure)…a nd why?Purification strategies•Protein stabilisation:–Integral membrane Proteins: Solubilisation–Purification at 4°C: reduces protease activity–Addition of protease inhibitors: commonly used are EDTA and PMSF (toxic) –Quickly load on first column after cell disruption and ultra centrifuge•Main impurities are removed first, lesser in the second or thirdstep•Max 5% impurities are acceptableIn general:Max 4 purification stepsNo steps with purification factor < 5No steps with < 30% yieldNo steps which last longer than one day and one night Chromatography •Separation material in columns, streamed by buffer →liquid chromatography HPLC: high pressure/performance liquid chromatographyFPLC: fast protein liquid chromatographyFPLC unitSeparation principles•Size:size exclusion chromatography (= gel filtration, = gel permeation chromatography) •Chargeanion or cation exchange chromatography; chromatofocusing •Hydrophobicityhydrophobic interaction chromatography (HIC)•Affinityaffinity chromatography•Solubilityammonium sulfate precipitation (non chromatographic, rel. imprecise)Size exclusion chromatography•Different pore sizes, depending on the size of the proteins •Separation is based in diffusion slow flow rate •Pressure sensitive materialsExample for a separation by gel filtrationNoteworthy about chromatographyGel filtration:limited sample volume: 2-3mllimited flow rate gel filtraion takes timedelutionof the sample by a factor of about 3low purificatopn factor: 3-6needs column buffer with high ionic strength: min 0.1 MIon exchage chromatography:To remember:Protein binding depents on electrostatic interactions with the column material.Strength of binding depents on pH and ionic strength of the buffer, the pI of the protein and the charge density on the column.In general:Technical easier than gel filtration: columns could be packed at the FPLC.Sample volume can be multiple times the column volume.Higher flowrates.Purification factor: 3-15Sample gets concentrated.Don’t use charged detergents!Ion exchage chromatographyBinding behaviour of proteins•pI= isoelectricpoint of the protein the pH value at which the posses no net charge •The pI determines the charge of the protein at a given pHpH > pI negative net charge pH < pIpositive net chargepIand protein separation on ion exchange columnsBut...•pIis not alone responsible for the binding behaviour of proteinsBinding is sometimes influenced by local charges and not by thenet charge of the proteinExample for a separation by ion exchange chromatographyMaterialsCarrier materials•Poly sugars–Sepharose (Agarose), Sephadex (Dextran), Sephacel (Cellulose) –Rare sugars can hardly be utilised by bacteria.–Low flow rates–Material changes it’s Volume depending on the ion ic strangth –Material settles over time•Beads–Polystyrol/Divinylbenzen beads with charge carrier added–Relative high flow rates–Good pressurestability, no compression–Hard charges could stress the protein•Beads and tentacle–Charges are linked with flexiblespacers to polymeric beads. This leadsto a soft binding of proteins despite of hard charges on the matrix –Relative high flow rates–Good pressure stability–High capaticity•Perfusion beads–Porous material, beads with chanels –Very big surface–Highly pressure stable–Very high flow ratesQuality of the separation。
CHM655/755 Experiment 8Purification of proteins by gel filtration chromatography Material:-Sephadex™ G-75-Tall/thin column-Tubing clams- 1 x TM buffer or other buffer systems dependent upon the target proteins to be separated.-Fraction collection tubes-Fraction collector-Erlenmeyer flasks and beakers-Parafilm-Marker pens-RulerProteins: lysozyme (14.7 kDa) and transferrin (75 kDa).Procedure:1.Determine the amount of resin to be used. Based on the information in the tableabove, 1 g of G-75 dried resin can produce 12-15 ml of slurry in ddH2O.2.Wash the resin extensively with ddH2O in a beaker. (Note: handle the resin gentlyto prevent breakage of the fragile beads.)3.Suspend the resin gently in ddH2O to a final volume that is twice the desiredpacked bed volume.4.Set up the column and make sure that the column is absolutely vertical. Close theoutlet at the bottom of the column.5.Place approximately one-tenth column volume of ddH2O into the empty column.6.Pour the resin slurry into the column in one continuous motion. Do it slowly andmake sure the resin settle down to the bottom without any air bubbles.7.Let the resin stand for a few minutes, and then open the outlet at the bottom of thecolumn to allow flow of ddH2O through the column.8.After the resin settles, close the outlet at the bottom of the column, and place acolumn flow adapter on the top of the resin bed. It is important for gel filtration that the top of the slurry is even all around and no any bubbles are trapped inside the slurry packed in the column. Make sure that the upper flow adapter is filled with ddH2O before putting it onto the column; otherwise, bubbles will be introduced into the column from the flow adapter.9.Before the sample is loaded to the column, it is necessary to equilibrate thecolumn with at least a 5 column volume of the buffer (1 x TM). Normally, the washing and elution buffers are the same. Carefully watch the washing buffer volume to avoid the column to be dried.10.Set the fraction collector to 30-40 drops in each tube, producing approximately 2ml of the eluate. Properly label the tubes for fraction collection.11.Once completion of the column washing, clamp the bottom line and load theprotein mixture gently (10 mg/ml). After the sample (0.5-1.5 ml) completely gets into the resin, load about 2 ml of the elution buffer slowly on the top of the resin bed, then connect the top line to the elution buffer reservoir. Unclamp the bottom line and allow factions to be collected.12.After completion of the collection, measure the O.D. value at 280 nm to obtainchromatographs of the separated proteins. The eluates do not need to be diluted for measurement and calculate the concentration based on the O.D. values. The samples then are placed at -20o C freezer for SDS-PAGE, western blot and other analyses.To test the properties of the column, it is a good practice to run a sample of 2mg/ml blue dextran on the newly poured column. Make a solution of 2mg/ml blue dextran in the chromatography buffer and filter to remove small, undissolved particles of blue dextran. Load the blue dextran in a volume of approximately 2% of the total column volume. Collect fractions using the automatic collector.Protein Absorbance Assay (280 nm)Considerations for useAbsorbance assays are fast and convenient, since no additional reagents or incubations are required. No protein standard need be prepared. The assay does not consume the protein. The relationship of absorbance to protein concentration is linear. Because different proteins and nucleic acids have widely varying absorption characteristics there may be considerable error, especially for unknowns or protein mixtures. Any non-protein component of the solution that absorbs ultraviolet light will interfere with the assay. Cell and tissue fractionation samples often contain insoluble or colored components that interfere. The most common use for the absorbance assay is to monitor fractions from chromatography columns, or any time a quick estimation is needed and error in protein concentration is not a concern. An absorbance assay is recommended for calibrating bovine serum albumin or other pure protein solutions for use as standards in other methods.PrincipleProteins in solution absorb ultraviolet light with absorbance maxima at 280 and 200 nm. Amino acids with aromatic rings are the primary reason for the absorbance peak at 280 nm. Peptide bonds are primarily responsible for the peak at 200 nm. Secondary, tertiary, and quaternary structure all affect absorbance, therefore factors such as pH, ionic strength, etc. can alter the absorbance spectrum.EquipmentIn addition to standard liquid handling supplies a spectrophotometer with UV lamp and quartz cuvette are required.ProcedureCarry out steps 1-4 (280 nm only) for a very rough estimate. Carry out all steps if nucleic acid contamination is likely.Warm up the UV lamp (about 15 min.)Adjust wavelength to 280 nmCalibrate to zero absorbance with buffer solution onlyMeasure absorbance of the protein solutionAdjust wavelength to 260 nmCalibrate to zero absorbance with buffer solution onlyMeasure absorbance of the protein solutionAnalysisUnknown proteins or protein mixtures. Use the following formula to roughly estimate protein concentration. Path length for most spectrometers is 1 cm.Concentration (mg/ml) = Absorbance at 280 nm divided by path length (cm.)Pure protein of known absorbance e the following formula for a path length of 1 cm. Concentration is in mg/ml, %, or molarity depending on which type coefficient is used.concentration = Absorbance at 280 nm divided by absorbance coefficientTo convert units, use these relationships:Mg protein/ml = % protein divided by 10 = molarity divided by protein molecular weightUnknowns with possible nucleic acid e the following formula to estimate protein concentration:Concentration (mg/ml) = (1.55 x A280) - 0.76 x A260)CommentsCold solutions can fog up the cuvette, while warm solutions can release bubbles and interfere with the readings. For concentrated solutions (absorbance greater than 2) simply dilute the solution.Absorbance coefficients of some common protein standards:Bovine serum albumin (BSA): 63Bovine, human, or rabbit IgG: 138Chicken ovalbumin: 70If the coefficient for a specific protein at a certain concentration is unknown, a standard curve is needed to accurately calculate the concentration of the protein.。
Protein PurificationHandbook 18-1132-29Edition ABBack toCollectionHiTrap, Sepharose, STREAMLINE, Sephadex, MonoBeads, Mono Q,Mono S, MiniBeads, RESOURCE, SOURCE, Superdex, Superose, HisTrap, HiLoad, HiPrep, INdEX, BPG, BioProcess, FineLINE, MabTrap, MAbAssistant, Multiphor, FPLC, PhastSystem and ÄKTA are trademarks of Amersham Pharmacia Biotech Limitedor its subsidiaries.Amersham is a trademark of Nycomed Amersham plcPharmacia and Drop Design are trademarks of Pharmacia & Upjohn Inc Coomassie is a trademark of ICI plcAll goods and services are sold subject to the terms and conditions of sale of the company within the Amersham Pharmacia Biotech group which supplies them. A copy of these terms and conditions of sale is available on request.© Amersham Pharmacia Biotech AB 1999-All rights reserved.Amersham Pharmacia Biotech ABSE-751 84 Uppsala SwedenAmersham Pharmacia Biotech UK Limited Amersham Place Little Chalfont Buckinghamshire England HP7 9NA Amersham Pharmacia Biotech Inc800 Centennial Avenue PO Box 1327 Piscataway NJ 08855 USAProtein Purification HandbookContents Introduction (7)Chapter 1Purification Strategies - A Simple Approach (9)Preparation (10)Three Phase Purification Strategy (10)General Guidelines for Protein Purification (12)Chapter 2 Preparation (13)Before You Start (13)Sample Extraction and Clarification (16)Chapter 3Three Phase Purification Strategy (19)Principles (19)Selection and Combination of Purification Techniques (20)Sample Conditioning (26)Chapter 4Capture (29)Chapter 5Intermediate Purification (37)Chapter 6Polishing (40)Chapter 7Examples of Protein Purification Strategies (45)Three step purification of a recombinant enzyme (45)Three step purification of a recombinant antigen binding fragment (49)Two step purification of a monoclonal antibody (54)One step purification of an integral membrane protein (57)Chapter 8Storage Conditions (61)Extraction and Clarification Procedures (62)Chapter 9Principles and Standard Conditions for Purification Techniques (73)Ion exchange (IEX) (73)Hydrophobic interaction (HIC) (79)Affinity (AC) (85)Gel filtration (GF) (88)Reversed phase (RPC) (92)Expanded bed adsorption (EBA) (95)IntroductionThe development of techniques and methods for protein purification has been an essential pre-requisite for many of the advancements made in biotechnology. This booklet provides advice and examples for a smooth path to protein purification. Protein purification varies from simple one-step precipitation procedures to large scale validated production processes. Often more than one purification step is necessary to reach the desired purity. The key to successful and efficient protein purification is to select the most appropriate techniques, optimise their performance to suit the requirements and combine them in a logical way to maximise yield and minimise the number of steps required.Most purification schemes involve some form of chromatography. As a result chromatography has become an essential tool in every laboratory where protein purification is needed. The availability of different chromatography techniques with different selectivities provides a powerful combination for the purification of any biomolecule.Recombinant DNA developments over the past decade have revolutionised the production of proteins in large quantities. Proteins can even be produced in forms which facilitate their subsequent chromatographic purification. However, this has not removed all challenges. Host contaminants are still present and problems related to solubility, structural integrity and biological activity can still exist. Although there may appear to be a great number of parameters to consider, with a few simple guidelines and application of the Three Phase Purification Strategy the process can be planned and performed simply and easily, with only a basic knowledge of the details of chromatography techniques.78Chapter 1Purification Strategies- a simple approachApply a systematic approach to development of a purification strategy. The first step is to describe the basic scenario for the purification. General considerations answer questions such as: What is the intended use of the product? What kind of starting material is available and how should it be handled? What are the purity issues in relation to the source material and intended use of the final product? What has to be removed? What must be removed completely? What will be the final scale of purification? If there is a need for scale-up, what consequences will this have on the chosen purification techniques? What are the economical constraints and what resources and equipment are available?Most purification protocols require more than one step to achieve the desired level of product purity. This includes any conditioning steps necessary to transfer the product from one technique into conditions suitable to perform the next technique. Each step in the process will cause some loss of product. For example, if a yield of 80% in each step is assumed, this will be reduced to only 20% overall yield after 8 processing steps as shown in Figure 1. Consequently, to reach the targets for yield and purity with the minimum number of steps and the simplest possible design, it is not efficient to add one step to another until purity requirements have been fulfilled. Occasionally when a sample is readily available purity can be achieved by simply adding or repeating steps. However, experience shows that, even for the most challenging applications, high purity and yield can be achieved efficiently in fewer than four well-chosen and optimised purification steps. Techniques should be organised in a logical sequence to avoid the need for conditioning steps and the chromatographic techniques selected appropriately to use as few purification steps as possible.Limit the number of steps in a purification procedure910Fig.1.Yields from multi-step purifications.PreparationThe need to obtain a protein, efficiently, economically and in sufficient purity and quantity, applies to every purification. It is important to set objectives for purity,quantity and maintenance of biological activity and to define the economical and time framework for the work. All information concerning properties of the target protein and contaminants will help during purification development. Some simple experiments to characterise the sample and target molecule are an excellent investment. Development of fast and reliable analytical assays is essential to follow the progress of the purification and assess its effectiveness. Sample preparation and extraction procedures should be developed prior to the first chromatographic purification step.With background information, assays and sample preparation procedures in place the Three Phase Purification Strategy can be considered.Three Phase Purification Strategy Imagine the purification has three phases Capture, IntermediatePurification and Polishing.In the Three Phase Strategy specific objectives are assigned to each step within the process:In the capture phase the objectives are to isolate, concentrate and stabilise the target product.During the intermediate purification phase the objective is to remove most of the bulk impurities such as other proteins and nucleic acids, endotoxins and viruses.In the polishing phase the objective is to achieve high purity by removing any remaining trace impurities or closely related substances.The selection and optimum combination of purification techniques for Capture,Intermediate Purification and Polishing is crucial to ensure fast method development, a shorter time to pure product and good economy.108060402012345678Number of steps 95% / step90% / step 85% / step 80% / step 75% / stepYield (%)The final purification process should ideally consist of sample preparation, including extraction and clarification when required, followed by three major purification steps, as shown in Figure 2. The number of steps used will always depend upon the purity requirements and intended use for the protein.Fig. 2.Preparation and the Three Phase Purification Strategy11Guidelines for Protein PurificationThe guidelines for protein purification shown here can be applied to any purification process and are a suggestion as to how a systematic approach can be applied to the development of an effective purification strategy. As a reminder these guidelines will be highlighted where appropriate throughout the following chapters.Define objectivesfor purity, activity and quantity required of final product to avoid over or under developing a methodDefine properties of target protein and critical impuritiesto simplify technique selection and optimisationDevelop analytical assaysfor fast detection of protein activity/recovery and to work efficientlyMinimise sample handling at every stageto avoid lengthy procedures which risk losing activity/reducing recovery Minimise use of additivesadditives may need to be removed in an extra purification step or may interfere with activity assaysRemove damaging contaminants earlyfor example, proteasesUse a different technique at each stepto take advantage of sample characteristics which can be used for separation (size, charge, hydrophobicity, ligand specificity)Minimise number of stepsextra steps reduce yield and increase time, combine steps logicallyKEEP IT SIMPLE!12Chapter 2PreparationBefore You StartThe need to obtain a protein, efficiently, economically and in sufficient purity and quantity, applies to any purification, from preparation of an enriched protein extract for biochemical characterisation to large scale production of a therapeutic recombinant protein. It is important to set objectives for purity and quantity, maintenance of biological activity and economy in terms of money and time. Purity requirements must take into consideration the nature of the source material, the intended use of the final product and any special safety issues. For example, it is important to differentiate between contaminants which must be removed and those which can be tolerated. Other factors can also influence the prioritisation of objectives. High yields are usually a key objective, but may be less crucial in cases where a sample is readily available or product is required only in small quantities. Extensive method development may be impossible without resources such as an ÄKTA™design chromatography system. Similarly, time pressure combined with a slow assay turnaround will steer towards less extensive scouting and optimisation. All information concerning properties of the target protein and contaminants will help during purification development, allowing faster and easier technique selection and optimisation, and avoiding conditions which may inactivate the target protein.Development of fast and reliable analytical assays is essential to follow the progress of the purification and assess effectiveness (yield, biological activity, recovery).Define objectivesGoal:To set minimum objectives for purity and quantity, maintenance of biological activity and economy in terms of money and time.Define purity requirements according to the final use of the product. Purity requirement examples are shown below.Extremely high > 99%Therapeutic use, in vivo studiesHigh 95- 99 %X-ray crystallography and most physico-chemicalcharacterisation methodsModerate < 95 %Antigen for antibody productionN-terminal sequencing13Identify 'key' contaminantsIdentify the nature of possible remaining contaminants as soon aspossible.The statement that a protein is >95% pure (i.e. target protein constitutes 95% of total protein) is far from a guarantee that the purity is sufficient for an intended application. The same is true for the common statement "the protein was homogenous by Coomassie™ stained SDS-PAGE". Purity of 95% may be acceptable if the remaining 5% consists of harmless impurities. However, even minor impurities which may be biologically active could cause significant problems in both research and therapeutic applications. It is therefore important to differentiate between contaminants which must be removed completely and those which can be reduced to acceptable levels. Since different types of starting material will contain different contaminant profiles they will present different contamination problems.It is better to over-purify than to under-purify.Although the number of purification steps should be minimised, thequality of the end product should not be compromised. Subsequent results might be questioned if sample purity is low and contaminants are unknown.Contaminants which degrade or inactivate the protein or interfere withanalyses should be removed as early as possible.The need to maintain biological activity must be considered at every stage during purification development. It is especially beneficial if proteases are removed and target protein transferred into a friendly environment during the first step.Economy is a very complex issue. In commercial production the time to market can override issues such as optimisation for recovery, capacity or speed. Robustness and reliability are also of great concern since a batch failure can have major consequences.It may be necessary to use analytical techniques targetted towards specific conta-minants in order to demonstrate that they have been removed to acceptable levels. 14Define properties of target protein and critical impurities Goal:To determine a 'stability window' for the target protein for easier selection and optimisation of techniques and to avoid protein inactivation during purification.Check target protein stability window for at least pH and ionic strength. All information concerning the target protein and contaminant properties will help to guide the choice of separation techniques and experimental conditions for purification. Database information for the target, or related proteins, may give size, isoelectric point (pI) and hydrophobicity or solubility data. Native one and two dimensional PAGE can indicate sample complexity and the properties of the target protein and major contaminants. Particularly important is a knowledge of the stability window of the protein so that irreversible inactivation is avoided. Itis advisable to check the target protein stability window for at least pH and ionic strength. Table 1 shows how different target protein properties can affect a purification strategy.Table 1.Protein properties and their effect on development of purification strategies. Sample and target protein properties Influence on purification strategyTemperature stability Need to work rapidly at lowered temperaturepH stability Selection of buffers for extraction and purificationSelection of conditions for ion exchange, affinity orreversed phase chromatographyOrganic solvents stability Selection of conditions for reversed phasechromatographyDetergent requirement Consider effects on chromatographic steps and the needfor detergent removal. Consider choice of detergent.Salt (ionic strength)Selection of conditions for precipitation techniques andhydrophobic interaction chromatographyCo-factors for stability or activity Selection of additives, pH, salts, buffersProtease sensitivity Need for fast removal of proteases or addition ofinhibitorsSensitivity to metal ions Need to add EDTA or EGTA in buffersRedox sensitivity Need to add reducing agentsMolecular weight Selection of gel filtration mediaCharge properties Selection of ion exchange conditionsBiospecific affinity Selection of ligand for affinity mediumPost translational modifications Selection of group specific affinity medium Hydrophobicity Selection of medium for hydrophobic interactionchromatography15Develop analytical assaysGoal:To follow the progress of a purification, to assess effectiveness (yield, biological activity, recovery) and to help during optimisation.Select assays which are fast and reliable.To progress efficiently during method development the effectiveness of each step should be assessed. The laboratory should have access to the following assays:• A rapid, reliable assay for the target protein• Purity determination• Total protein determination• Assays for impurities which must be removedThe importance of a reliable assay for the target protein cannot be over- emphasised. When testing chromatographic fractions ensure that the buffers used for separation do not interfere with the assay. Purity of the target protein is most often estimated by SDS-PAGE, capillary electrophoresis, reversed phase chromatography or mass spectrometry. Lowry or Bradford assays are used most frequently to determine the total protein.The Bradford assay is particularly suited to samples where there is a high lipid content which may interfere with the Lowry assay.For large scale protein purification the need to assay for target proteins and critical impurities is often essential. In practice, when a protein is purified for research purposes, it is too time consuming to identify and set up specific assays for harmful contaminants. A practical approach is to purify the protein to a certain level, and then perform SDS-PAGE after a storage period to check for protease cleavage. Suitable control experiments, included within assays forbio-activity, will help to indicate if impurities are interfering with results.Sample Extraction and Clarification Minimise sample handlingMinimise use of additivesRemove damaging contaminants earlyDefinition:Primary isolation of target protein from source material.Goal:Preparation of a clarified sample for further purification. Removal of particulate matter or other contaminants which are not compatible with chromatography.16The need for sample preparation prior to the first chromatographic step is dependent upon sample type. In some situations samples may be taken directly to the first capture step. For example cell culture supernatant can be applied directly to a suitable chromatographic matrix such as Sepharose™ Fast Flow and may require only a minor adjustment of the pH or ionic strength. However, it is most often essential to perform some form of sample extraction and clarification procedure.If sample extraction is required the chosen technique must be robust and suitable for all scales of purification likely to be used. It should be noted that a technique such as ammonium sulphate precipitation, commonly used in small scale, may be unsuitable for very large scale preparation. Choice of buffers and additives must be carefully considered if a purification is to be scaled up. In these cases inexpensive buffers, such as acetate or citrate, are preferable to the more complex compositions used in the laboratory. It should also be noted that dialysis and other common methods used for adjustment of sample conditions are unsuitable for very large or very small samples.For repeated purification, use an extraction and clarification techniquethat is robust and able to handle sample variability. This ensures areproducible product for the next purification step despite variability instarting material.Use additives only if essential for stabilisation of product or improvedextraction. Select those which are easily removed. Additives may need tobe removed in an extra purification step.Use pre-packed columns of Sephadex™ G-25 gel filtration media, forrapid sample clean-up at laboratory scale, as shown in Table 2.Table 2.Pre-packed columns for sample clean-up.Pre-packed column Sample volume Sample volume Code No.loading per run recovery per runHiPrep™Desalting 26/10 2.5 -15 ml7.5 - 20 ml17-5087-01HiTrap Desalting0.25 - 1.5 ml 1.0 - 2.0 ml17-1408-01Fast Desalting PC 3.2/100.05 - 0.2 ml0.2 - 0.3 ml17-0774-01PD-10 Desalting 1.5 - 2.5 ml 2.5 - 3.5 ml17-0851-01 Sephadex G-25 gel filtration media are used at laboratory and production scale for sample preparation and clarification of proteins >5000. Sample volumes of up to 30%, or in some cases, 40% of the total column volume are loaded. In a single step, the sample is desalted, exchanged into a new buffer, and low molecular weight materials are removed. The high volume capacity, relative insensitivity to sample concentration, and speed of this step enable very large sample volumes to be processed rapidly and efficiently. Using a high sample volume load results in a separation with minimal sample dilution (approximately 1:1.4). Chapter 8 contains further details on sample storage, extraction and clarification procedures.17Sephadex G-25 is also used for sample conditioning i.e. rapid adjustment of pH, buffer exchange and desalting between purification steps.Sephadex G-25 gel filtrationFor fast group separations between high and low molecular weight substances Typical flow velocity 60 cm/h (Sephadex G-25 SuperFine, Sephadex G-25 Fine), 150 cm/h (Sephadex G-25 Medium).If large sample volumes will be handled or the method scaled-up in the future, consider using STREAMLINE™ expanded bed adsorption. This technique is particularly suited for large scale recombinant protein and monoclonal antibody purification. The crude sample containing particles can be applied to the expanded bed without filtration or centrifugation. STREAMLINE adsorbents are specially designed for use in STREAMLINE columns. Together they enable the high flow rates needed for high productivity in industrial applications of fluidised beds. The technique requires no sample clean up and so combines sample preparation and capture in a single step. Crude sample is applied to an expanded bed STREAMLINE media. Target proteins are captured whilst cell debris, cells, particulate matter, whole cells, and contaminants pass through. Flow is reversed and the target proteins are desorbed in the elution buffer.STREAMLINE (IEX, AC, HIC)For sample clean-up and capture direct from crude sample.STREAMLINE adsorbents are designed to handle feed directly from both fermentation homogenate and crude feedstock from cell culture/fermentation at flow velocities of 200 - 500 cm/h, according to type and application.Particle size: 200 µmNote:cm/h: flow velocity (linear flow rate) = volumetric flow rate/cross sectional area of column.18Chapter 3Three Phase Purification StrategyPrinciplesWith background information, assays, and sample preparation and extraction procedures in place the Three Phase Purification Strategy can be applied (Figure 3). This strategy is used as an aid to the development of purification processes for therapeutic proteins in the pharmaceutical industry and is equally efficient as an aid when developing purification schemes in the research laboratory.Fig. 3.Preparation and the Three Phase Purification Strategy.Assign a specific objective to each step within the purification process.In the Three Phase Strategy a specific objective is assigned to each step. The purification problem associated with a particular step will depend greatly upon the properties of the starting material. Thus, the objective of a purification step will vary according to its position in the process i.e. at the beginning for isolation of product from crude sample, in the middle for further purification of partially purified sample, or at the end for final clean up of an almost pure product.The Three Phase Strategy ensures faster method development, a shorter time to pure product and good economy.In the capture phase the objectives are to isolate, concentrate and stabilise the target product. The product should be concentrated and transferred to an environment which will conserve potency/activity. At best, significant removal of other critical contaminants can also be achieved.19During the intermediate purification phase the objectives are to remove most of the bulk impurities,such as other proteins and nucleic acids, endotoxins and viruses.In the polishing phase most impurities have already been removed except for trace amounts or closely related substances. The objective is to achieve final purity.It should be noted that this Three Phase Strategy does not mean that all strategies must have three purification steps. For example, capture and intermediate purification may be achievable in a single step, as may intermediate purification and polishing. Similarly, purity demands may be so low that a rapid capture step is sufficient to achieve the desired result, or the purity of the starting material may be so high that only a polishing step is needed. For purification of therapeutic proteins a fourth or fifth purification step may be required to fulfil the highest purity and safety demands.The optimum selection and combination of purification techniques for Capture, Intermediate Purification and Polishing is crucial for an efficient purification process.Selection and Combination ofPurification TechniquesMinimise sample handlingMinimise number of stepsUse different techniques at each stepGoal:Fastest route to a product of required purity.For any chromatographic separation each different technique will offer different performance with respect to recovery, resolution, speed and capacity. A technique can be optimised to focus on one of these parameters, for example resolution, or to achieve the best balance between two parameters, such as speed and capacity.A separation optimised for one of these parameters will produce results quite different in appearance from those produced using the same technique, but focussed on an alternative parameter. See, for example, the results shown on page 49 where ion exchange is used for a capture and for a polishing step.20Select a technique to meet the objectives for the purification step. Capacity,in the simple model shown, refers to the amount of target protein loaded during purification. In some cases the amount of sample which can be loaded may be limited by volume (as in gel filtration) or by large amounts of contaminants rather than the amount of the target protein.Speed is of the highest importance at the beginning of a purification where contaminants such as proteases must be removed as quickly as possible. Recovery becomes increasingly important as the purification proceeds because of the increased value of the purified product. Recovery is influenced by destructive processes in the sample and unfavourable conditions on the column. Resolution is achieved by the selectivity of the technique and the efficiency of the chromatographic matrix to produce narrow peaks. In general, resolution is most difficult to achieve in the final stages of purification when impurities and target protein are likely to have very similar properties.Every technique offers a balance between resolution, speed, capacity and recovery and should be selected to meet the objectives for each purification step. In general, optimisation of any one of these four parameters can only be achieved at the expense of the others and a purification step will be a compromise. The importance of each parameter will vary depending on whether a purification step is used for capture, intermediate purification or polishing. This will steer the optimisation of the critical parameters, as well as the selection of the most suitable media for the step.Proteins are purified using chromatographic purification techniques which separate according to differences in specific properties, as shown in Table 3. Table 3.Protein properties used during purification.Protein property TechniqueCharge Ion exchange (IEX)Size Gel filtration (GF)Hydrophobicity Hydrophobic interaction (HIC),reversed phase (RPC)Biorecognition (ligand specificity)Affinity (AC)Charge, ligand specificity or hydrophobicity Expanded bed adsorption (EBA) follows theprinciples of AC, IEX or HIC21。
His-tag融合蛋白的纯化The following protocols is for His Bind resinHis Bind Purification1) Express and harvest cells. Resuspend cells in Binding Buffer. Lyse cells.2) Prepare resin (~20- 25 ml?) in column - wet column and frit, transfer resin slurry to column. Wash with water (3 volume), Charge Buffer (5 volumn), Binding Buffer (3 volume).3) Load column with cell lysate, at a rate of ~10 column volumn per hour.4) Wash column (10 X volumn) with Binding Buffer5) Wash column (6 X volumn) with Wash Buffer6) Elute protein (6 X volume) with Elute Buffer (strip buffer can also be used to elute). Alternatively elute first with lower concentration of imidazole at 0.25 M.7) T o store column, wash with Strip Buffer (3 X volumn) and stored in Strip Buffer at 4 °C.1X BufferPurification in Denaturing Condition (protein in inclusion body)1) Same as before.2) Centrifuge to collect inclusion body. Decant supernatant, resuspend in Binding Buffer (sonicate if necessary), centrifuged. Repeat until all trapped proteins are released.3) Decant supernatant, resuspend in Binding Buffer + 6 M guanidine or Urea. Incubate on ice for 1 hour to dissolve.4) Centrifuged at 39,000 g for 20 min. Filter supernatant (0.45 micron membrane).5) Load onto column. Purification same as before, except that all buffer contain denaturant, and lower imidazole concentration in Wash Buffer (20 mM) and Elute Buffer (~300mM) (dilute Wash and Elute Buffer with Binding Buffer). Alternatively, you can wash and elute the protein without imidazole at low pH (wash at pH 6.5, elute at pH5.9 or pH4.5 if the protein failed to elute at the hight pH)Regeneration of Column- when flow rate is slow and column resin doesn't turn blue-green when charged1) Wash with 2 vol 6 M guanidine-HCl, then 3 vol water.2) Wash with 1 vol 2% SDS.3) Wash with 1 vol each of 25%, 50% and 75% ethanol, then 5 vol of 100% ethanol, followed by 1 vol each of 75%, 50% and 25% ethanol.4) Wash with 1 vol water, then 5 volume 100 mM EDTA (pH 8.0).5) Wash with 3 vol water, then 3 vol 20% ethanol.6) Store at 4 °C.Note:1) Do not use ßME, DTT or EDTA in buffer.2) Higher amount of imidazole may be used in the wash buffer (100 mM) but some protein may be eluted.3) You can also elute the protein with salt concentration gradient or using lower pH.4) It's sometimes useful to introduce a couple of glycines between your protein and the His-tag which allow the his-tag to be fully exposed for effective binding to resin (if your vector doesn't have a short linker sequence between the HIs-tag and your protein that is).His-tag融合蛋白纯化中Ni柱的使用Materials and reagentsProcedureGST融合蛋白纯化方法Abstract: Many people have vented out frustration over insoluble GST-fused proteins. This is a protocol for enzymatically active soluble GST-fused proteins. All GST-fused proteins are rendered soluble with this technique though enzyme activitiy can range from 30-90%.Materials and Reagents1.STE Buffer10 mM Tris-HCl, pH 8.01 mM EDT A150 mM NaCl2.Lysozyme solution10 mg/ml in water (make fresh)3.PBS4.Elution Buffer50 mM Tris.Cl, pH 9.020 mM GSH5.10% Sarkosyl in STE Buffer6.10% Triton X-100 in STE Buffer7. 1 M DTT8.100 mM IPTGProcedureDay 11.Set up an overnight culture in 50 ml 2XTY with 150 mg/ml of ampicillin.Day 21.Seed 5 ml of overnight culture to 500 ml 2XTY with 150 mg/ml of ampicillin.2.Grow at 37o C to an A600 of 0.6 to 0.8.3.Induce with 0.1 mM to 2 mM of IPTG. Grow for 3 hr at 37o C or grow overnight at roomtemperature.Lower IPTG concentrations and lower growing temperatures tend to produce greater solubility at the expense of yield.4.Pellet cells by centrifuging at 3000 g, 4o C for 10 min. Decant media and resuspend cellsin 30 ml ice-cold PBS to wash. Transfer to a 40-ml Oak Ridge tube and centrifuge at 3000 g, 4o C for 10 min. Decant PBS.5.This is a convenient point to stop and to store pellets at -80o C. Else continue to lysecells.6.Thaw pellet on ice if cells are frozen else proceed to the next step.7.Resuspend pellet in 10 ml of ice cold STE Buffer.8.Add 100 ml of freshly prepared lyozyme solution, incubate on ice for 15 min. Justbefore sonication, add 100 ml of 1 M DTT and 1.4 ml of 10% Sarkosyl. Mix thoroughly by inversion and sonicate for a total time of 1 min.9.Centrifuge 16,000 rpm for 20 min on the SS34 rotor to pellet debris. Transfersupernatant to a 50-ml conical tube and discard the pellet. Add 4 ml of 10% Triton X-100 and top up with STE Buffer to 20 ml. The effective concentration of Sarkosyl and Triton X-100 will be 0.7% and 2% respectively. Incubate at room temperature for30 min.10.Pour the lysate to 1 ml bed of prepared Glutathione Sepharose in PBS. Incubat e atroom temperature for 30 min to 1 hr with agitation.To prepare the 50% slurry, shake up the media and pipette 2 ml to a 50 mltube. Fill to 50 ml with PBS, invert tube a few times. Centrifuge to 2000 rpmon a swing bucket centrifuge then switch off. Carefully suck off PBS andresuspend beads with 1 ml of PBS.11.Wash the beads with 3 X 50 ml of PBS. Finally resuspend in 5 ml of PBS. Pour to adispo-column. Wash the 50-ml conical tube with an additional 5 ml of PBS. Pool withthe first 5 ml in the dispo-column.To wash, use the same centrifugation technique for preparing the beads. When transferring beads to column, do not pipette but pour. The beads tend to stickto pipette tips.12.If desired, elute with 10 x 1 ml fractions of Elution Buffer. Determine desired fractionswith SDS PAGE.ReferenceFrangioni and Neel.Anal. Biochem. 210, 179-187 (1993)酵母中GST融合蛋白纯化方法∙ 5 ml overnight culture of your favorite yeast in your favorite medium.∙Inoculate 50 ml and grow 30o C shaking O/N until OD600 = 0.8 to 1.2. For SCD cultures use 1/500 and 1/1500 dilutions.∙Optional: add alpha-factor to 2.5 µM. Continue shaking at 30o C for 60 min.∙Add 1 M NaN 3 to 10 mM (final concentration) and move cultures to ice. Everything must remain cold from here on out.∙Spin cells 3 K, 10 min at 4o C.∙Discard supe and resuspend cells with 0.5 ml 10 mM NaN3. Transfer to 1.5 ml microfuge tube (not autoclaved).∙Spin 3 K, 10 min at 4o C. Alternatively spin 8 K, 1 min at room temperature.∙Discard supe. Optional: freeze pellet at -80o C.∙Resuspend in 1 ml of cold 10 mM NaN3.∙Measure OD600. Adjust volumes so that there are an equal number of cells in each sample. A total OD600 of 30 per sample is best.∙Wash with 1 ml of Lysis Buffer.∙Spin 3 K 10 min at 4o C and discard supe.∙Resuspend in 400 ul Lysis Buffer.∙Add a scoop of glass beads to a 0.5 ml PCR tube. Transfer cell lysate to the PCR tube∙Vortex 1 min, 4X. Keep samples cold between vortexing.∙Poke a hole in the bottom of the tube and spin cell lysate in a new microfuge tube 1.5 K or 500 X g, 10 min., 4o C.∙Transfer liquid from bottom tube into a new microfuge tube.∙Spin again 1.5 K, 10 min, 4o C and again transfer liquid into a new microfuge tube.∙Add Triton X-100 to 1.5 % and rock for 60 min at 4o C.∙Spin (3 K, 10 min, 4o C) and transfer the supe to a new microfuge tube.∙Remove 30 ul of liquid and add 30 ul 2X SDS PAGE Sample buffer. This will reflect protein content before Glutathione purification.∙T o the remaining liquid, add 100 ul 40 % slurry of Glutathione beads and mix at 4o C for 2 h (overnight is usually fine). Glutathione beads should be prewashed 3 X with PBS and 1 X with lysis buffer before resuspending as a 40 % slurry in lysis buffer.∙Wash glutathione beads five times with PBS, 1 % Triton X-100, 300 mM NaCl at RT. Spin 2 K, 5 min, at room temperature. Rock sample for 5 min between washes. Change tubes after the first wash to reduce nonspecific binding to the tube itself.∙Resuspend in SDS-PAGE Sample buffer.o Alternatively elute 3 times with 1-2 column volumes of 5-10 mM reduced glutathione, 50 mM Tris pH 8, and mix with 6X SDS-PAGE sample bufferbefore stripping the beads with SDS-PAGE sample buffer.∙Heat to 100o C for 10 min. Then store at -20o C. Protein is ready to be run on SDS-PAGE Gel.NOTESThanks to Paul DiBello and Jiyoung Cha for their refinements of this protocol.* Indicates a correction from an eariler version of the protocol.1. For lysis in the presense of GDP and GTP I use a final concentration of 10 uM GDP or 20 uM GTPgammaS and a final concentration of 3 mM MgCl2 in the lysis buffer.2. You can substitute protease inhibitor cocktail (Sigma P8215) for individual protease inhibitors (AEBSF, leupeptin, pepstatin, benzamidine, aprotinin).3. For lysis to determine phosphorylation, you may wish to add more phosphatase inhibitors (in addition to beta-glycerolphosphate and Na-o-vanadate) to the lysis buffer, or you may wish to omit phosphatase inhibitors altogether:50mM Na-M-Vanadate 200ml 0.5mM100mM2 ml 10mMNa-pyrophosphate2mg/ml Phosvitin 10µl 1µg/ml。