核酸序列分析ppt课件
- 格式:ppt
- 大小:9.66 MB
- 文档页数:18


第4章核酸序列分析了解:1.DNA携带的两类遗传信息。
2.DNA与RNA序列分析的常见内容及相关数据库和工具。
3.ORF与CDS的区别。
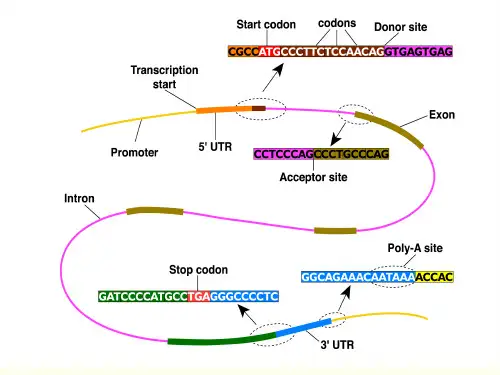
4.原核基因和真核基因启动子的结构。
5.原核和真核的基因结构。
6.lncRNA的研究现状。
熟悉:1.限制性核酸内切酶的命名规则,II型限制酶的特点。
2.重复序列依重复次数和组织形式的分类。
3.基因识别的三大类方法。
4.miRNA及其靶基因预测的方法和工具。
掌握:1.CpG岛的概念及其识别依据和判别标准。
2.mRNA选择性剪接的产生机制。
3.解决问题的思路。
4.查找数据库和分析工具的方法。
5.学习数据库与分析工具使用方法的策略。
4.1引言“龙生龙,凤生凤,老鼠的儿子会打洞!”1“种瓜得瓜,种豆得豆。
”“爹矬矬一个,娘矬矬一窝。
”“一母生九子,连母十个样。
”“龙生九子各不同。
”“天下乌鸦一般黑。
”这些都是大家耳熟能详的谚语。
不管是天上飞的、地上跑的、水里游的,还是能动的、不能动的,它们的后代都和它们非常相像,但却也会有少许的差异。
这些现象大家都已司空见惯,所以可能没有啥感觉。
但仔细想想,你就会发现大自然的奇妙所在。
当然,对于生物专业的人来说,这个就没什么奇怪的了,因为我们都知道分子生物学的中心法则(The central dogma of molecular biology):DNA转录成RNA,RNA翻译成蛋白质。
蛋白质执行特定的生物功能从而决定最终的表型,而DNA则携带着最原始的决定个体性状的遗传信息,RNA主要参与遗传信息的表达和调控。
在各种生物中,A、C、G、T/U都是构成DNA和RNA核酸序列的基本组分。
仅仅这么四种碱基怎么可能构建出缤纷多彩的大千世界呢?其秘诀就在于四种核苷酸的排列顺序。
就像搭积木一样,通过不同的排列组合我们可以构建出不同的形状。
类似于二进制中运用一连串的0和1以及英文字母表中运用26个不同的字母来表达信息,基因所包含的信息来自于4中不同核苷酸沿DNA 分子的排列顺序。




核酸序列分析在生物学领域中,核酸序列分析是一项重要的研究工具,它可以帮助科学家们理解生物体内的基因组结构和功能。
通过分析核酸序列,我们可以揭示基因的组合方式、基因在不同物种之间的演化关系以及基因与疾病之间的关联。
本文将介绍核酸序列分析的基本步骤和常用方法,并探讨它在生物研究中的应用。
一、核酸序列分析的基本步骤1. 数据收集与清洗:首先,我们需要获取相关的核酸序列数据。
这些数据可以来自于公共数据库(如GenBank、ENSEMBL等)或实验室内部的测序项目。
收集到的数据可能存在噪声或错误,所以我们需要对数据进行清洗和筛选,以保证分析的准确性。
2. 序列比对:接下来,我们需要将不同样本的核酸序列进行比对。
序列比对是核酸序列分析的核心步骤之一,它可以帮助我们发现序列之间的相似性和差异性。
常用的序列比对算法包括Smith-Waterman算法和Needleman-Wunsch算法等。
3. 序列注释:在比对完成后,我们可以根据已知的功能注释信息来对序列进行注释。
注释可以告诉我们该序列可能的编码蛋白质的功能、寻找潜在的基因等。
4. 比对结果分析:通过分析比对结果,我们可以了解到序列的保守区域和变异区域。
保守区域可能是功能区域,例如编码蛋白质的区域,变异区域可能涉及到物种之间的进化差异或突变相关的功能。
5. 结果可视化:最后,我们需要将分析的结果进行可视化呈现。
通过可视化,我们可以更直观地理解数据,并对进一步实验设计或研究方向提出建议。
二、核酸序列分析的常用方法1. 比对工具:常用的核酸序列比对工具包括BLAST、ClustalW和MAFFT等。
BLAST(基本局部比对序列工具)是一种快速的局部比对算法,它能够快速地找到序列之间的相似性。
ClustalW和MAFFT则更适用于多序列比对,它们可以比较多个序列之间的相似性和差异性。
2. 注释工具:常用的核酸序列注释工具包括NCBI的Entrez、ENSEMBL和UniProt等。

生物化学中的核酸序列分析生物化学是研究生命现象与生理功能的科学,而核酸是构成生命的分子之一,它们在生物体内扮演着重要的角色。
核酸是由核苷酸单元组成的长链,其中DNA是一个双螺旋分子,可以储存生物遗传信息,而RNA则可以转录DNA的信息并参与蛋白质合成。
在生物研究中,对核酸序列的分析非常重要。
通过对DNA序列的分析,可以推测出蛋白质编码信息并预测基因功能;而对RNA序列的分析,则可以了解基因的表达和调控。
本文将从分子生物学和生物信息学的角度来探讨核酸序列分析。
1. PCR扩增与测序分析PCR(聚合酶链式反应)是一种常用的分子生物学技术,可以从少量的DNA或RNA样品中扩增出目标片段,为进一步的分析提供足够的材料。
PCR过程中需要用到一组引物,其可以通过生物信息学分析DNA序列寻找到设计合适的引物。
PCR扩增得到的产物可以进一步进行测序分析,最常用的测序方式为Sanger测序技术。
此技术基于DNA链延伸过程中的dNTP和ddNTP的竞争关系,通过荧光信号和电泳进行测序。
测序结果可以通过生物信息学工具进行比对、序列注释和统计分析。
2. 基因功能预测高通量基因组测序技术的出现,导致了大量未知基因序列的暴增。
对于这些基因序列的功能预测,通常需要先进行同源比对。
同源比对基于多序列比对的原理,将物种间已知的方向同源序列,与未知序列比对,寻找到相似的序列区域,从而对未知序列的基因功能进行推测。
同源比对时,需要注意序列的物种来源和序列的质量。
不同物种间的序列可能在不同位置发生突变,导致序列的比对不准确;若序列存在较多的突变,也可能会影响比对结果。
因此,如何选择合适的工具和参数进行同源比对很关键。
同时,基因家族和重复序列也可能会干扰比对结果,因此需要进行筛除和过滤。
3. RNA测序与转录组分析RNA测序技术可以获得全基因组水平的转录信息,从而了解基因的表达状态和调控机理。
RNA测序通常经过文库构建和深度测序等多个步骤。


