产品生产放行审核记录
- 格式:doc
- 大小:35.00 KB
- 文档页数:1
成品放行审核制度1、目的:按照cGMP 规范要求,建立本公司产品放行的审核制度。
2、范围:本公司生产的所有产品的放行。
3、职责3.1生产车间主管指定有资格的生产人员按本程序审核批生产记录,交质量部审核和存档。
3.2检测中心主管指定有资格的检验人员按本程序审核批检验记录,交质量部审核和存档。
3.3质量部质量受权人按本程序审核执行批的所有记录,决定产品的放行与否。
4、程序4.1生产车间对批生产记录的审核4.1.1在一批产品生产过程完成后,生产车间应由有资格的生产人员对批生产记录进行第一轮审核。
审核内容包括但不限于以下方面:·准确性:所用记录是否为最新批准的版本;配料、称重是否准确;所有计算,包括物料平衡检查和收率计算是否正确。
·完整性:所有生产工序是否完成和记录;所有中间体检测项目是否检测和记录;批记录有无缺页、损坏;填写是否完全,是否有遗漏;是否有被操作人员遗漏的偏差。
·符合性:批生产记录填写是否符合要求,修改是否规范,是否有难辩认的更改;所有生产工序是否有操作人、复核人签名。
所用原料规格是否正确,投料比是否在规定范围内;工艺参数是否在规定范围内,有无偏差;中间体检测结果是否符合质量标准,有无超标或超趋势结果;物料平衡和收率是否在规定范围内,有无偏差;包装材料和标签是否正确,使用数量与包装数量是否一致;所有已发现的偏差是否已在批生产记录上作了备注,是否都已报告和评估;对关键的偏差是否做了调查,并附有调查报告;若有涉及该批的变更,是否有批准文件。
4.1.2以上审核完成后,生产车间审核人应在批生产记录的审核单上签名、署日期,然后交给质量部审核和存档。
4.2检测中心对批检验记录的审核4.2.1检测中心检测完该批产品的所有样品后,应由有资格的检验人员对批检验记录进行第一轮审核。
审核内容包括但不限于以下方面:·准确性:所用记录是否为最新批准的版本;样品和标准品称量和稀释是否准确;所有计算公式和计算是否正确。

Process audit checklistBDH256Change Level:Audit Date:Auditor:NO.评价evaluate1.11.21.31.4NO.评价evaluation2.12、设计和过程FMEA/design and process FMEA does the actual material and production flow follow the process flowchart?DFMEA 的零件编号、修订版本、原始日期和审定日期是否正确?does the DFMEA have the correct Part#, Revision Level, Orig. Date and Rev. Date?does the floor plan identify the following :all required assembly, process and inspection stations?locations for all raw material, work in process(WIP) and finished product?Is the flowcharts lastest and identified with date?记录/comments记录/commentsPart Name:Part Number :文 件 要 求/document questions是否有最新版平面布置图?生产过程中,材料和产品流动是否与过程流程图一致?is there a dated copy of the current floor plan readily available?操 作 要 求/implementation/process questions文 件 要 求/document questionsDFMEA (含有设计职责)/DFMEA(if supplier is design responsible)平面布置图上是否有所有装配、生产和检测岗位、原材料、半成品、成品的区域划分?流程图是否是最新版本的并标注有日期?Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number:3、控制计划/Control PlanProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :4、 检验和试验/Inspection and TestingProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :5、产品规范与图纸/Product Specifications & DrawingsProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :6、过程监控与操作指导书/Process Monitoring & Operator InstructionsProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :7、 Handling and storageProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :8、包装与发运规范/Parts Packaging/Shipping SpecificationsProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :9、异常处理Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :11、检测量具的评价/Gage & Check Fixture EvaluationProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :12、产能验证/Line Speed & Capacity VerifiedProcess audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :过程审核表Process audit checklistBDH256Change Level:Audit Date:Auditor:Part Name:Part Number :第 21 页,共 21 页。


产品放行审核管理制度一、引言产品放行审核是指根据相关规定和标准,对生产制造、经销销售的产品进行审核,确认其符合相关法规和标准要求,具备安全性和质量可靠性,方可放行上市销售。
产品放行审核管理制度是企业为保障产品质量和安全性,规范产品放行审核活动并确保审核过程的合法、透明和公正性而建立的一套制度和流程。
本文将围绕产品放行审核管理制度展开详细阐述。
二、产品放行审核的重要性1、保障产品质量和安全性。
产品放行审核是保障产品质量和安全性的重要环节,只有经过审核确认符合相关标准和法规要求的产品方可上市销售,从而降低产品质量安全风险,保护消费者权益。
2、维护企业声誉。
产品放行审核严格管理,有助于企业提高产品质量和合规水平,确保产品符合法规,从而维护企业的良好声誉和市场形象。
3、推动企业可持续发展。
严格执行产品放行审核管理制度,有利于企业提升管理水平和竞争力,增强产品质量控制和安全监管能力,推动企业实现可持续发展。
三、产品放行审核管理制度的实施1、制度建立(1)建立产品放行审核管理制度编写组织,并确定具体责任人员。
(2)明确制度编写的目的、范围和内容,建立完善的审核管理制度框架。
(3)结合企业业务特点和产品特性,确定产品放行审核相关流程、方法和要求。
2、审核流程(1)审核申请:企业申请产品放行审核,提交相关资料和样品。
(2)资质审查:审核人员对企业资质和产品信息进行审查,确保企业具备产品生产销售资质和产品信息真实准确。
(3)检测检验:产品放行审核需要进行产品检测和抽样检验,确保产品合格。
(4)审核决定:审核人员根据审核结果做出是否放行的决定。
(5)审核报告:将审核结果以及审核意见记录在产品放行审核报告中,确保审核结果的真实可靠。
3、审核方法(1)文件审核:审核资料的真实性和完整性,确保资料齐全准确。
(2)现场审核:对生产、质量控制等环节进行现场检查,确保符合标准和要求。
(3)检测检验:对产品进行抽样检测,确保产品质量达到标准。

产品放行单检验报告背景介绍产品放行单是指在生产加工过程中,对最终产品进行检验并确认合格后,才可正式放行的单据。
它是质量管理体系中非常重要的一环,用于确保产品质量符合标准要求,以保障消费者权益和企业形象。
检验过程产品放行单的制作和填写需要经过以下几个关键步骤:1. 产品取样在进行检验之前,必须从生产线中取得样品。
样本应代表整个批次产品的特征,并有代表性。
2. 定义检验指标根据产品的性质和相关法规标准,确定需要进行检验的指标和要求。
例如,食品安全检验可能包括卫生指标、添加剂限量等。
3. 进行实验室测试将样本送至实验室进行严格的检测。
实验室应具备权威认可的资质,并严格按照标准化操作程序进行测试。
4. 分析检测结果根据实验室的测试结果,对样本进行数据分析和解读,确认产品是否符合要求。
5. 填写放行单如果产品经过检验合格,负责检验的人员将填写放行单。
放行单通常包括产品信息、检验结果、检验人员签名、检验日期等。
6. 审核和批准经过检验的放行单需要由相关质量管理人员进行审核和批准。
审核人员应核实检验结果是否符合标准要求,并确保填写准确无误。
7. 存档完成审核和批准后,放行单将被归档保存,以备将来追溯和查阅。
重要性与意义产品放行单作为质量管理的重要环节,具有重要的意义和作用:1. 保证产品质量通过产品的严格检验,能够确保产品符合质量标准和要求,从而保障消费者的权益和安全。
2. 提高客户满意度产品放行单能够记录产品的全过程检验情况,使得消费者能够更加信任和满意企业的产品,提高企业的形象和声誉。
3. 优化质量管理流程产品放行单能够记录检验数据和结果,为质量管理人员提供宝贵的数据支持,帮助企业优化和改进质量管理流程,提高效率和准确性。
4. 法律和合规要求在某些行业中,如食品、医药等,产品放行单是法律和合规的要求。
企业必须依法进行严格的检验,并保留相应的检验报告。
结语产品放行单检验报告是确保产品质量合格的关键文件,通过严格的检验流程和文件记录,能够提高企业的质量管理水平和客户满意度。

产品质量审核记录单日期:_____________________________产品名称:_____________________________供应商名称:_____________________________批次号:_____________________________质量审核人员:_____________________________审核内容:1.原料质量评估:-对原料的质量进行评估,包括外观、气味、颜色等方面的检查,确保原料符合相应的标准和要求。
-对原料进行抽样检测,采用适当的检测方法和设备,检测原料的各项指标,确保原料的质量合格。
-对产品的生产工艺进行审核,包括生产过程中的各项操作流程、设备的选择和使用等。
-检查生产工艺是否符合相关的法规、标准和要求,确保生产过程的合法性和可行性。
3.产品质量检测:-对产品进行全面的质量检测,包括外观、尺寸、功能、性能等方面的检测。
-采用适当的检测方法和设备,检测产品的各项指标,确保产品的质量符合相应的标准和要求。
-对工厂的生产环境进行审核,包括生产车间、仓库、设备等方面的检查。
-检查工厂的环境是否符合相关的法规、标准和要求,确保工厂的生产环境符合卫生和安全的要求。
-对产品质量相关的文件记录进行审核,包括原材料采购记录、生产工艺记录、产品检测记录等。
-检查文件记录是否齐全、准确和完整,确保质量相关的信息可以被有效地追溯和管理。
审核结论:1.符合要求:-原料质量评估合格,生产工艺符合要求,产品质量检测合格,工厂环境符合要求,文件记录齐全、准确和完整。
-审核人员可以确认该产品的质量符合相关的标准和要求,可以放行该批次产品。
2.不符合要求:-原料质量评估不合格,生产工艺不符合要求,产品质量检测不合格,工厂环境不符合要求,文件记录不齐全、准确和完整。
-审核人员应对不符合要求的问题提出整改意见,并要求供应商进行相应的整改和改进,直到问题得到解决后方可放行该批次产品。
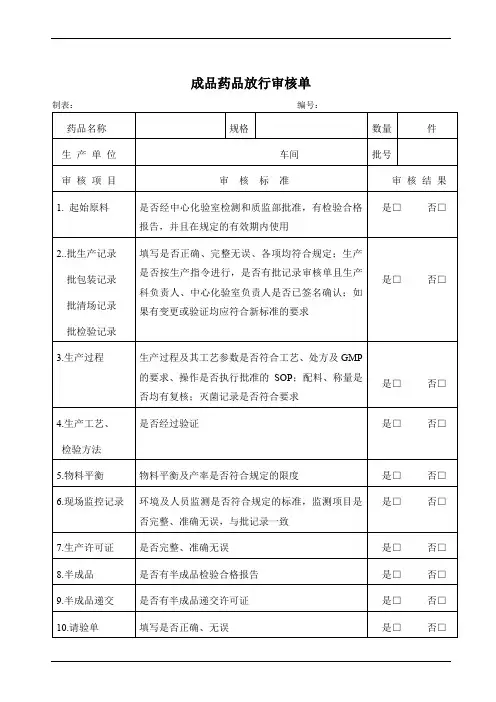
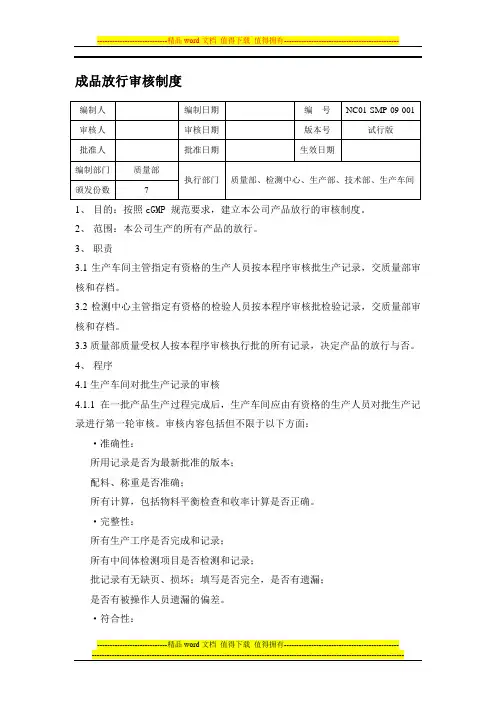
成品药品放行审核单 制表: 编号: 药品名称 规格 数量 件 生 产 单 位 车间 批号 审 核 项 目 审 核 标 准 审 核 结 果 1. 起始原料 是否经中心化验室检测和质监部批准,有检验合格报告,并且在规定的有效期内使用 是□ 否□
2..批生产记录 批包装记录 批清场记录 批检验记录 填写是否正确、完整无误、各项均符合规定;生产是否按生产指令进行,是否有批记录审核单且生产科负责人、中心化验室负责人是否已签名确认;如果有变更或验证均应符合新标准的要求
是□ 否□
3.生产过程 生产过程及其工艺参数是否符合工艺、处方及GMP的要求、操作是否执行批准的SOP;配料、称量是否均有复核;灭菌记录是否符合要求 是□ 否□
4.生产工艺、 检验方法 是否经过验证 是□ 否□
5.物料平衡 物料平衡及产率是否符合规定的限度 是□ 否□ 6.现场监控记录 环境及人员监测是否符合规定的标准,监测项目是否完整、准确无误,与批记录一致 是□ 否□
7.生产许可证 是否完整、准确无误 是□ 否□ 8.半成品 是否有半成品检验合格报告 是□ 否□ 9.半成品递交 是否有半成品递交许可证 是□ 否□ 10.请验单 填写是否正确、无误 是□ 否□ 11.偏差处理 如有偏差是否执行偏差处理管理规程并符合要求 是□ 否□ 12.变更处理 如有变更是否执行变更处理规程并符合要求 是□ 否□ 13.成品取样 是否执行批准的取样规程并符合要求 是□ 否□ 14.成品检验 是否执行批准的检验操作规程 是□ 否□ 15.成品报告 是否有成品检验合格报告单并检验合格 是□ 否□ 16.成品包装质量 是否对包装质量、印刷内容及有效期打印进行检验,并有合格的结果 是□ 否□
17.质量评价 药品及其生产是否符合注册要求、质量标准和GMP要求 是□ 否□ 审核人 审核日期 年 月 日 结论 □符合规定 □不符合规定
批准人 生效日期 年 月 日 结论 批放行 □ 复验 □ 拒绝放行□ 备注
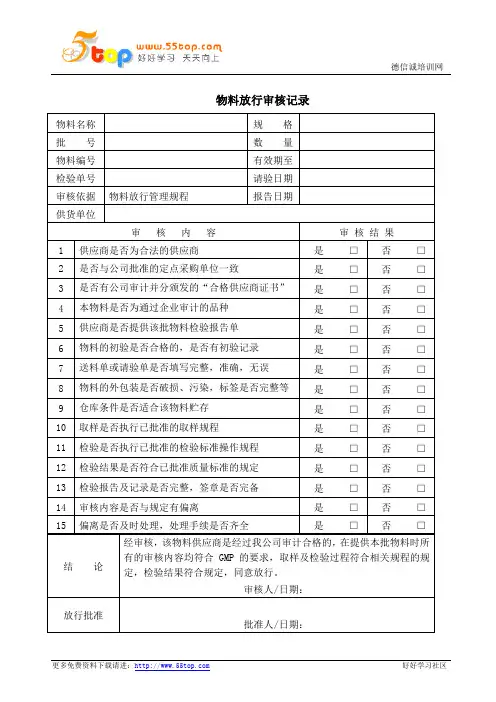
产品生产放行审核记录
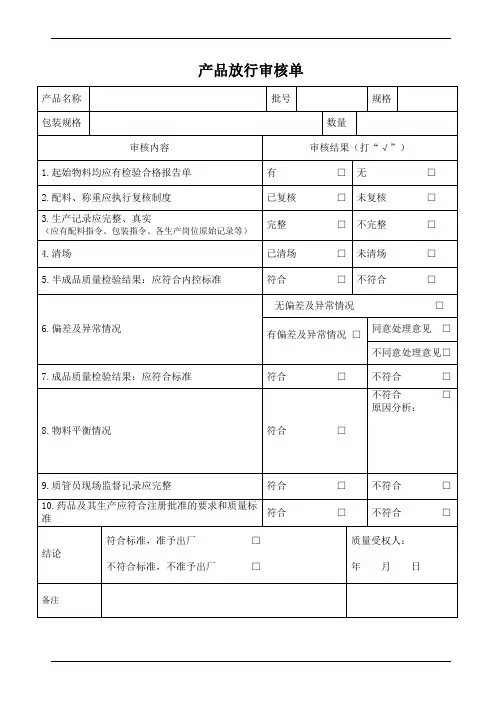
产品名称 批号 规格
包装规格 数量
审核内容 审核结果(打“√”)
生
产情况 1.首件产品经过检测 □ 是 □ 不是 2. 主要生产工艺经过验证 □ 是 □ 不是 3.生产环境条件、设备符合规范要求 □符合 □不符合
4.生产记录及时、完整、真实,可追溯
□ 是 □ 不是
5.生产记录经主管负责人审核、签名
□ 是 □ 不是
审核结论 □ 合格,批准放行。 质量受权人:
年 月 日
□ 不合格,不准放行。
备注