保护碱基选择
- 格式:doc
- 大小:56.00 KB
- 文档页数:4

各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH , 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer 完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。

各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments(oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg 已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。

PCR设计引物时酶切位点的保护碱基引物设计是PCR实验的关键步骤之一,引物的好坏会直接影响到PCR反应的成功与否。
而在引物设计过程中,酶切位点的保护碱基是需要考虑的重要因素之一在PCR实验中,引物的作用是指定PCR反应的放大区域,并提供启动位点供聚合酶结合。
一般情况下,引物至少需要包含一段特定的DNA序列,以便与目标序列互补配对。
在引物设计过程中,选择合适的酶切位点是十分必要的。
酶切位点是指位于特定DNA序列上的限制酶可以识别并切割的区域。
酶切位点的选择通常需要考虑如下几个方面:1.切割效果:选择切割效果好的酶切位点可以提高PCR反应的特异性和灵敏度。
经典的选择是选择一种具有4-6个碱基的酶切位点,并且该位点在引物中间的位置。
这可以有效防止酶切位点的保护碱基对PCR反应的影响。
2.特异性:引物需要选择适合的酶切位点,以确保只有目标序列被放大,而不包括其他与之相关的非特异性序列。
因此,在选择酶切位点时应尽量避免与其他非特异性序列存在相似性。
3.引物长度:引物长度的选择也与酶切位点相关。
如果引物长度过短,可能会导致酶切位点过于靠近PCR反应产物的端点,从而使切割效果不佳。
因此,在引物设计时,应选择适当的引物长度,以保证酶切位点的保护碱基不会对PCR反应产物的生成产生不利影响。
酶切位点的保护碱基是指在特定的DNA序列上,通过选择相应的碱基来避免受到酶切的影响。
常见的保护碱基有甲基化碱基、磷酸化碱基以及接上阻断扩增的非互补碱基等。
1.甲基化碱基:将酶切位点中的一些碱基进行甲基化处理,可以有效地阻止特定酶的切割作用。
甲基化碱基可以通过DNA甲基转移酶进行甲基化修饰。
2.磷酸化碱基:磷酸化碱基是在引物设计过程中添加磷酸基团的方法,通过给酶切位点添加一个磷酸基团来阻断酶的切割作用。
3.非互补碱基:为了阻断酶切位点的切割作用,可以在酶切位点的周围引入一个与其不互补的碱基序列。
这样可以阻断酶的结合和切割。
总的来说,选择合适的酶切位点和保护碱基对PCR实验的成功至关重要。
保护碱基
限制性内切酶识别特定的DNA序列,除此之外,酶蛋白还要占据识别位点两边的若干个碱基,这些碱基对内切酶稳定的结合到DNA双链并发挥切割DNA作用是有很大影响的,被称为保护碱基。
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
添加原则
1、添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
2、添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,
添加几个保护碱基,是有数据可以参考的,见附表。
3、添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC
含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
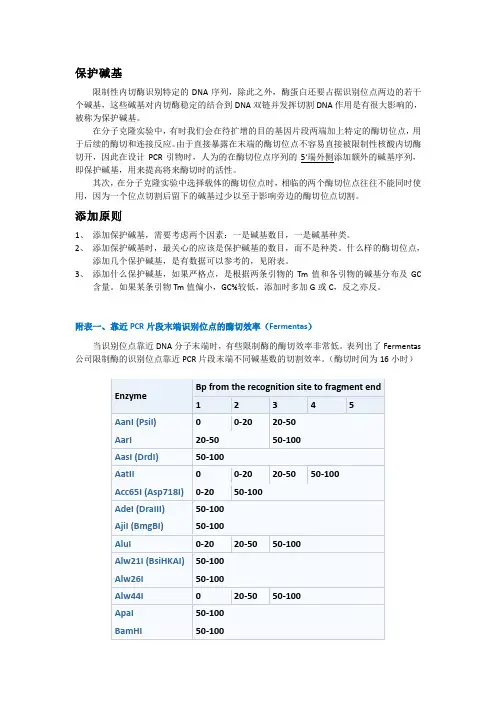
附表一、靠近PCR片段末端识别位点的酶切效率(Fermentas)
当识别位点靠近DNA分子末端时,有些限制酶的酶切效率非常低。
表列出了Fermentas 公司限制酶的识别位点靠近PCR片段末端不同碱基数的切割效率。
(酶切时间为16小时)
2、保护碱基列表(BioLabs)。

各种酶切位点的保护碱基引物设计必看酶切位点是指特定的序列,酶可以识别并在该位置切割DNA分子。
这些位点的特异性使得酶在分子生物学中广泛应用于DNA片段的定位和切割。
然而,在一些实验中,我们可能需要保护酶切位点周围的碱基,以免酶切,并且只在特定的位置引导酶切。
因此,保护碱基引物的设计对于实验的成功非常重要。
以下是保护碱基引物设计的一些建议。
首先,保护碱基引物的设计需要考虑引物的长度。
引物的长度通常为18到30个碱基,具体的长度需要根据实验的需求和酶切位点周围的序列特征来确定。
引物的长度应足够长,以确保引物和靶序列的特异性,但不应过长,以免引物形成二级结构或与非特异性位点结合。
其次,保护碱基引物的设计需要考虑引物的碱基组成。
在设计引物时,建议尽量避免引物中出现酶切位点周围的碱基序列,以防止酶的误切。
例如,如果我们希望保护酶切位点周围的AATTC序列,可以设计一个引物,其中没有AATTC序列。
同时,引物的碱基组成应尽量避免多聚核苷酸或含有GC碱基的片段,以防止引物之间的结合或引物与非特异靶序列的结合。
此外,保护碱基引物的设计需要考虑引物的特异性。
在设计引物时,建议使用特异性的引物序列,以确保引物只与目标酶切位点结合。
可以通过使用生物信息学工具,如BLAST,来验证引物的特异性。
引物的特异性还可以通过调整引物的长度和碱基组成来进一步提高。
最后,保护碱基引物的设计需要考虑引物的热力学性质。
引物的热力学性质包括引物的熔解温度(Tm值)和引物之间的配对。
引物的Tm值与引物的碱基组成、长度和引物与靶序列之间的碱基配对相关。
可以使用在线工具,如NEB的Tm计算器,来计算引物的Tm值,并对不同的引物进行比较。
此外,引物之间的配对可以通过设计引物的末端序列来调整,例如末端的碱基配对或非配对等。
总结起来,保护碱基引物的设计需要考虑引物的长度、碱基组成、特异性和热力学性质。
通过合理设计引物,可以保护酶切位点周围的碱基,并在特定位置引导酶切,为实验的成功提供有力的保障。
克隆PCR产物的方法之一,是在PCR产物两端设计一定的限制酶切位点,经酶切后克隆至用相同酶切的载体中。
但实验证明,大多数限制酶对裸露的酶切位点不能切断。
必须在酶切位点旁边加上一个至几个保护碱基,才能使所定的限制酶对其识别位点进行有效切断。
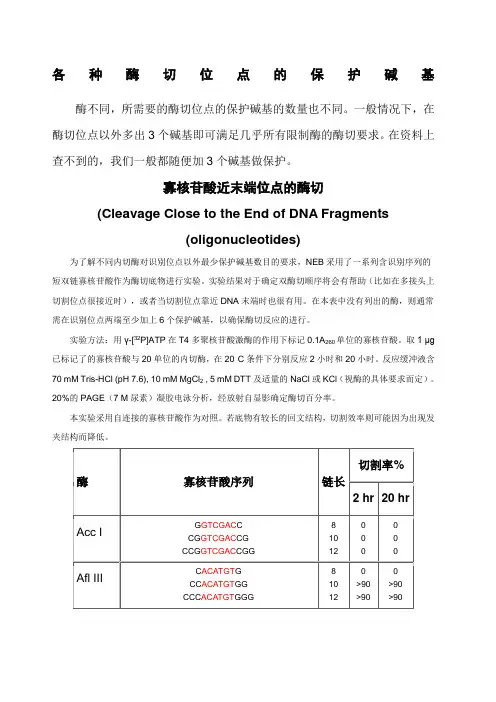
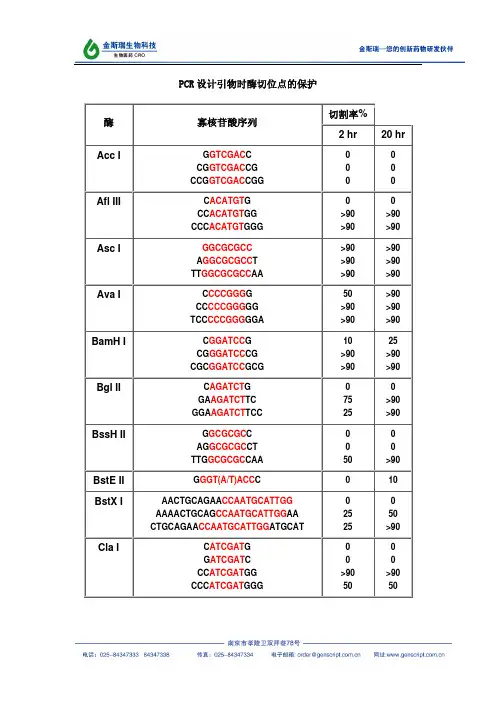
有研究者使用了15种限制酶,分别比较了各种限制酶在其酶切位点旁边分别加0、1、2、3个保护碱基后的切断情况。
结果显示,基本上所有限制酶,在其酶切位点旁边加上3个以上的保护碱基后,可以对其酶切位点进行有效切断。
一般来讲,在酶切位点前加入两个GC碱基,因为如果保护碱基为A T的话,保护碱基在PCR 产物的末端,A T之间只有两个氢键,结合力差,容易在末端产生单链,这样的话限制性内切酶就无法作用。
其实加保护碱基的多少,是具体情况具体讨论,比如HindIII、BamHI等就得有三个保护碱基,少了一个就无法切动。
注释:
1.如果要加在序列的5‘端,就在酶切位点识别碱基序列(红色)的5’端加上相应的碱基(黑色),相同如果要在3‘端加保护碱基,就在酶切位点识别碱基序列(红色)的3’端加上相应的碱基(黑色)。
2.切割率:正确识别并酶切的效率
3. 加保护碱基时最好选用切割率高时加的相应碱基。
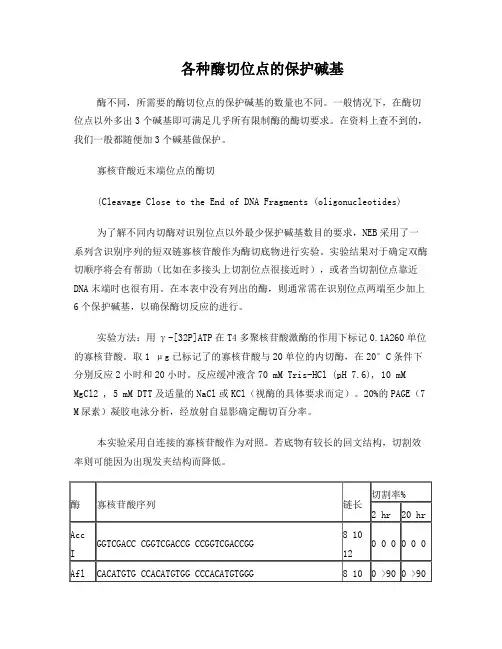
各种酶切位点的保护碱基酶不同,所需要的酶切位点的保护碱基的数量也不同。
一般情况下,在酶切位点以外多出3个碱基即可满足几乎所有限制酶的酶切要求。
在资料上查不到的,我们一般都随便加3个碱基做保护。
寡核苷酸近末端位点的酶切(Cleavage Close to the End of DNA Fragments (oligonucleotides)为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
实验方法:用γ-[32P]ATP在T4多聚核苷酸激酶的作用下标记0.1A260单位的寡核苷酸。
取1 μg已标记了的寡核苷酸与20单位的内切酶,在20°C条件下分别反应2小时和20小时。
反应缓冲液含70 mM Tris-HCl (pH 7.6), 10 mM MgCl2 , 5 mM DTT及适量的NaCl或KCl(视酶的具体要求而定)。
20%的PAGE(7 M尿素)凝胶电泳分析,经放射自显影确定酶切百分率。
本实验采用自连接的寡核苷酸作为对照。
若底物有较长的回文结构,切割效率则可能因为出现发夹结构而降低。
2.双酶切的问题参看目录,选择共同的buffer。
其实,双酶切选哪种buffer是实验的结果,takara公司从1979年开始生产限制酶以来,做了大量的基础实验,也积累了很多经验,目录中所推荐的双酶切buffer完全是依据具体实验结果得到的。
有共同buffer的,通常按照常规的酶切体系,在37℃进行同步酶切。
但BamH I在37℃下有时表现出star活性,常用30℃单切。
两个酶切位点相邻或没有共同 buffer的,通常单切,即先做一种酶切,乙醇沉淀,再做另一种酶切。

Cleavage Close to the End of DNA Fragments (linearized vector)Linearized vectors were incubated with the indicated enzymes (10 units/µg) for 60 minutes at the recommended incubation temperature and NEBuffer for each enzyme. Following ligation and transformation, cleavage efficiencies were determined by dividing the number of transformants from the digestion reaction by the number obtained from religation of the linearized DNA (typically 100-500 colonies) and subtracting from 100%. "Base Pairs from End" refers to the number of double-stranded base pairs between the recognition site and the terminus of the fragment; this number does not include the single-stranded overhang from the initial cut. Since it has not been demonstrated whether these single-stranded nucleotides contribute to cleavage efficiency, 4 bases should be added to the indicated numbers when designing PCR primers. Average efficiencies were rounded to the nearest whole number; experimental variation was typically within 10%. The numbers in parentheses refer to the number of independent trials for each enzyme tested (from Moreira, R. and Noren, C. (1995), Biotechniques, 19, 56-59).Note: As a general rule, enzymes not listed below require 6 bases pairs on either side of their recognition site to cleave efficiently.New England Biolabs Technical Literature - Updated 03/05/2004To test the varying requirements restriction endonucleases have for the number of bases flanking their recognition sequences, a series of short, double-stranded oligonucleotides that contain the restriction endonuclease recognition sites (shown in red) were digested. This information may be helpful when choosing the order of addition of two restriction endonucleases for a double digest (a particular concern when cleaving sites close together in a polylinker), or when selecting enzymes most likely to cleave at the end of a DNA fragment.The experiment was performed as follows: 0.1 A260 unit of oligonucleotide was phosphorylated using T4 polynucleotide kinase and -[32P] ATP. 1 µg of 5´ [32P]-labeled oligonucleotide was incubated at 20°C with 20 units of restriction endonuclease in a buffer containing 70 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT and NaCl or KCl depending on the salt requirement of each particular restriction endonuclease. Aliquots were taken at 2 hours and 20 hours and analyzed by 20% PAGE (7 M urea). Percent cleavage was determined by visual estimate of autoradiographs.As a control, self-ligated oligonucleotides were cleaved efficiently. Decreased cleavage efficiency for some of the longer palindromic oligonucleotides may be caused by the formation of hairpin loops.。

酶切位点保护碱基表:PRC引物保护碱基的设计首先要明确什么是保护碱基限制性内切酶识别特定的DNA序列,除此之外,酶蛋白还要占据识别位点两边的若干个碱基,这些碱基对内切酶稳定的结合到DNA双链并发挥切割DNA作用是有很大影响的,被称为保护碱基。
添加保护碱基的目的在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
但实验证明,大多数限制酶对裸露的酶切位点不能切断。
必须在酶切位点旁边加上一个至几个保护碱基,才能使所定的限制酶对其识别位点进行有效切断。
因此在设计PCR引物时,为保护5` 端外加的内切酶识别位点,人为地在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高酶切时的活性,使酶切完全。
其次,在分子克隆实验中选择载体的酶切位点时,相临的两个酶切位点往往不能同时使用,因为一个位点切割后留下的碱基过少以至于影响旁边的酶切位点切割。
添加保护碱基的原则添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的。
一般情况下,普通的内切酶只加入两个保护碱基,其内切反应就可以正常进行;而有一类,仅仅只加入两个保护碱基,其内切反应就不能正常进行,这是因为内切酶不能正常结合DNA段上。
如NdeI就属这类,需要加入至少6个保护碱基,常用的HindIII也要三个。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
为了解不同内切酶对识别位点以外最少保护碱基数目的要求,NEB采用了一系列含识别序列的短双链寡核苷酸作为酶切底物进行实验。
实验结果对于确定双酶切顺序将会有帮助(比如在多接头上切割位点很接近时),或者当切割位点靠近DNA末端时也很有用。
在本表中没有列出的酶,则通常需在识别位点两端至少加上6个保护碱基,以确保酶切反应的进行。
在分子克隆实验中,有时我们会在待扩增的目的基因片段两端加上特定的酶切位点,用于后续的酶切和连接反应。
由于直接暴露在末端的酶切位点不容易直接被限制性核酸内切酶切开,因此在设计PCR引物时,人为的在酶切位点序列的5‘端外侧添加额外的碱基序列,即保护碱基,用来提高将来酶切时的活性。
添加保护碱基,需要考虑两个因素:一是碱基数目,一是碱基种类。
添加保护碱基时,最关心的应该是保护碱基的数目,而不是种类。
什么样的酶切位点,添加几个保护碱基,是有数据可以参考的,见附表。
添加什么保护碱基,如果严格点,是根据两条引物的Tm值和各引物的碱基分布及GC含量。
如果某条引物Tm值偏小,GC%较低,添加时多加G或C,反之亦反。
保护碱基列表。
各种酶切位点的保护碱基引物设计必看酶切位点的保护碱基引物设计在分子生物学领域中起着至关重要的作用。
它们是研究者在酶切实验中必不可少的工具,用于保护酶切位点周围的碱基,以避免酶的切割作用。
本文将介绍保护碱基引物设计的一般原则和具体步骤,并探讨一些常见的问题和注意事项。
保护碱基引物设计的一般原则如下:1.引物长度:保护碱基引物的长度通常为15-25个碱基对。
2.引物序列:引物应根据酶切位点的序列设计。
为了确保引物的特异性,通常将酶切位点和其周围的碱基考虑在内。
3.引物组成:引物的核苷酸组成应考虑碱基的GC含量,以保持引物的稳定性。
通常,GC含量高于50%的引物更稳定。
4.引物末端修饰:引物的末端修饰可以提高引物与目标DNA的亲和性,并增加引物的稳定性。
常用的末端修饰包括磷酸化和胺基修饰等。
保护碱基引物设计的步骤如下:1.获取酶切位点序列:首先,需要获取目标DNA序列中待保护的酶切位点的序列。
2.引物设计:根据酶切位点的序列设计引物。
引物的长度通常为15-25个碱基对。
为了提高特异性,可以考虑在引物序列中加入一些限制性内切酶无法识别的碱基。
3.引物末端修饰:根据需要选择引物的末端修饰方式,例如磷酸化和胺基修饰等。
4.引物的合成:完成引物设计后,可以委托专业的生物科技公司进行引物的合成。
确保引物的纯度和质量。
在进行保护碱基引物设计时,还需注意一些常见的问题和注意事项:1.引物特异性:在设计引物时,要确保引物与目标DNA的序列具有高度特异性,以避免引物与非目标区域的杂交。
2.引物的稳定性:引物的稳定性对于酶切实验的成功至关重要。
在设计引物时,要尽量选择稳定的引物序列,例如具有较高GC含量的引物。
3.引物纯度和质量:为了保证引物的质量和稳定性,引物的合成必须由专业的生物科技公司进行。
确保引物的纯度高,无杂质。
4.引物的浓度和稀释:在使用引物进行酶切实验时,要合理确定引物的浓度和稀释倍数,以保证实验的成功。
总之,保护碱基引物设计是分子生物学研究中不可或缺的一部分。
常用酶切位点序列和保护碱基引言在分子生物学和遗传工程领域,酶切位点序列和保护碱基是非常重要的概念。
酶切位点序列指的是DNA或RNA上特定的核苷酸序列,这些序列可以被特定的酶识别并切割。
保护碱基则是指在实验过程中采取措施来保护DNA或RNA上特定的核苷酸,使其不被酶切割。
本文将对常用的酶切位点序列和保护碱基进行详细介绍,包括其定义、常见的酶切位点序列、如何选择合适的保护碱基等内容。
酶切位点序列定义酶切位点序列是指DNA或RNA分子上具有一定规律性、可以被特定的限制性内切酶识别并结合从而发挥催化作用的核苷酸序列。
这些限制性内切酶通常能够识别4-8个核苷酸,并在识别到相应的位点后将DNA或RNA分子切割成片段。
常见的酶切位点序列1.EcoRI: 5’-GAATTC-3’,3’-CTTAAG-5’2.HindIII: 5’-AAGCTT-3’,3’-TTCGAA-5’3.BamHI: 5’-GGATCC-3’,3’-CCTAGG-5’4.XhoI: 5’-CTCGAG-3’,3’-GAGCTC-5’5.NotI: 5’-GCGGCCGC-3’,3’-CGCCGGCG-5’这些酶切位点序列是常用的限制性内切酶的识别序列,它们在分子生物学实验中被广泛应用。
通过将DNA或RNA与特定的限制性内切酶一起反应,可以实现DNA或RNA的特定部位切割。
保护碱基定义保护碱基是指在实验过程中采取措施来保护DNA或RNA上特定的核苷酸,使其不被酶切割。
这种保护通常通过对特定的碱基进行修饰或使用化学试剂来实现。
如何选择合适的保护碱基选择合适的保护碱基需要考虑以下几个因素: 1. 酶切位点序列:首先要了解所使用的限制性内切酶的酶切位点序列,以确定需要保护的碱基。
2. 保护方法:根据实验需求和实验条件选择合适的保护方法。
常见的保护方法包括使用化学修饰剂修饰碱基、使用特殊的核苷酸引物或引入特定的修饰基团等。
3. 保护效果:选择的保护碱基应能够有效地阻止限制性内切酶与目标位点结合并发挥催化作用。
各种酶切位点的保护碱基酶切位点是指酶在DNA或RNA分子中特定的位置识别并切割的区域。
在分子生物学研究中,酶切位点的保护碱基是进行引物设计的重要参考依据之一、本文将介绍各种酶切位点的保护碱基以及在引物设计中的应用。
1.核酸酶A切割位点保护碱基核酸酶A(RNase A)是一种特定的核酸酶,能够将单链RNA切割为5'-磷酸核酸和3'-核磷酸。
核酸酶A识别和切割位点的保护碱基主要为对尿嘧啶核苷酸(Uridine,U)和鸟嘌呤核苷酸(Adenine,A)。
具体来说,核酸酶A主要作用于RNA链上U和A的周围碱基,特别是位于U或A的下一个碱基。
在引物设计中,需要考虑核酸酶A切割位点的保护碱基,以避免产生无法预测的酶切割产物。
特别是在设计RNA引物时,需要避免在酶切位点附近出现U和A的保护碱基。
2.核酸酶T1切割位点保护碱基核酸酶T1(RNase T1)是一种特定的核酸酶,能够识别并切割由磷酸鸟苷(Guanosine,G)形成的RNA链。
核酸酶T1在RNA链上识别和切割位点的保护碱基为G。
具体来说,核酸酶T1主要作用于G的下一个碱基。
在引物设计中,需要考虑核酸酶T1切割位点的保护碱基,以避免产生无法预测的酶切割产物。
特别是在设计RNA引物时,需要避免在酶切位点附近出现G的保护碱基。
3.限制性内切酶切割位点保护碱基限制性内切酶是一类广泛应用于DNA分子生物学研究的酶,其识别和切割DNA分子中的特定序列。
限制性内切酶识别和切割位点的保护碱基由酶自身的特异性决定。
每一种限制性内切酶都有其特定的酶切位点保护碱基要求。
一般来说,酶切位点的保护碱基主要存在于酶切位点的上下游碱基中。
在引物设计中,需要考虑限制性内切酶切割位点的保护碱基,以避免引物和酶切位点之间存在相互作用而导致切割不完全或无法切割的情况。
总而言之,在引物设计中,需要考虑各种酶切位点的保护碱基,以提高引物的特异性和稳定性。
根据不同酶的切割特点和要求,设计合适的引物序列可以避免酶切位点的保护碱基产生干扰,保证实验结果的准确性。
保护碱基名词解释保护碱基是保护在有机合成或生物合成中容易受到碱性条件下水解或其他负面影响的碱基,以确保化学反应成功进行并保持结构完整。
保护通常使用特定化合物对碱基进行保护,待需要时再去掉保护基团。
一、保护碱基的原因1. 防止碱性水解:碱性条件下,碱基会被破坏,需要通过保护来防止这一点。
2. 避免竞争反应:保护碱基使其在需要的时间内保持完整,避免在反应过程中参与竞争反应。
3. 促进立体选择性反应:通过保护碱基来控制反应过程,可以实现立体选择性反应,得到需要的产物。
4. 促进分子识别:保护碱基可以将某些官能团与其他官能团区分开来,促进分子识别。
5. 稳定反应条件:有时添加保护基团可以传导反应中所需的能量,从而有助于稳定反应条件。
二、常用的碱基保护基团1. 酰基:如酰氯、酯、酰亚胺等,以保护酚、醇、胺等碱基。
2. 甲基、乙基:以保护醛、酮、羧酸等碱基。
3. 芳香基:以保护氨基或羟基等碱基。
4. 三氟乙酰基、戊二酰亚胺基:以保护酰胺或胺等碱基。
5. 丁二酰基:以保护氨苯酮等碱基。
6. 叔丁基二甲基硅基:以保护配体、氨基和羟基等。
三、去除碱基保护基团的方法1. 酸性条件:常见的酸性去保护方法包括用氢氟酸、硫酸、苯磺酸等酸性物质。
2. 碱性条件:用钠或钾水银齐、氢氧化钾等碱性物质可以去除酰保护基团。
3. 其他方法:还有氢化还原、光化学、试剂氧化等去保护方法。
四、保护碱基在有机合成中的应用1. 保护羟基:在多数羟基和苯酚发生取代反应后进行去保护,以防止产生重排反应,同时可以使其在齐平反应中发挥特殊作用。
2. 保护氨基和胺基:将原先的胺基氨基化配合物合成保护合成物,多用于化合物的合成中。
3. 保护羰基:可以通过保护羰基保护对下攻击基团的反应活性。
4. 保护烯基:通过保护烯基可以调整烯酮在取代反应中的反应性,避免侧反应的发生。
总之,保护碱基是有机合成中必不可少的方法之一,通过保护可以使反应更加选择性,得到更加纯净的产物,为药物合成、有机物合成等提供了重要的帮助。