化药4类申报资料清单
- 格式:doc
- 大小:57.50 KB
- 文档页数:2

附件一:新药(化学药品)申报资料项目第一部分综述资料1.新药名称(包括通用名、化学名、英文名、汉语拼音。
凡新制定的名称,应说明依据),选题的目的与依据,国内外有关该品研究现状或生产、使用情况的综述。
2.研制单位研究工作的综述。
3.产品包装、标签设计样稿。
4.使用说明书样稿。
第二部分药学资料5.原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。
6.确证化学结构或组分的试验资料及文献资料。
7.质量研究工作的试验资料及文献资料。
包括理化性质、纯度检查、溶出度、含量测定等。
8.质量标准草案及起草说明,并提供标准品或对照品。
9.临床研究用的样品及其检验报告书(申请;临床时报送)或生产的样品3~5批及其检验报告书(申请生产时报送)。
10.稳定性研究的试验资料及文献资料。
11.产品包装材料及其选择依据。
第三部分药理毒理资料12.主要药效学试验资料及文献资料。
13.一般药理研究的试验资料及文献资料。
14.急性毒性试验资料及文献资料。
15.长期毒性试验资料及文献资料。
16.局部用药毒性研究的试验资料及文献资料,全身用药的过敏性、溶血性、血管刺激性等试验资料及文献资料。
17.复方制剂中多种组分药效、毒性。
药代动力学相互影响的试验资料及文献资料。
18.致突变试验资料及文献资料。
19.生殖毒性试验资料及文献资料。
20.致癌试验资料及文献资料。
21.依赖性试验资料及文献资料。
22.药代动力学试验资料及文献资料。
第四部分临床资料23.供临床医生参阅的药理、毒理研究及文献的综述。
24.临床研究计划及研究方案。
25.临床研究总结资料(包括知情同意书、伦理委员会批准件)。
说明1.新药(化学药品)申请临床研究时报送附件一项目1~24;申请生产时报送附件一项目1~25。
2.放射性新药申报资料的要求详见所附《放射性新药申报资料项目及说明》,其各类放射性新药参照同类别化学药品的要求报送资料。
3.国内外尚未上市的新药,国外机构在我国申请注册者,可以申报在国外完成的研究资料,但应按我国的研究资料项目要求归类整理。
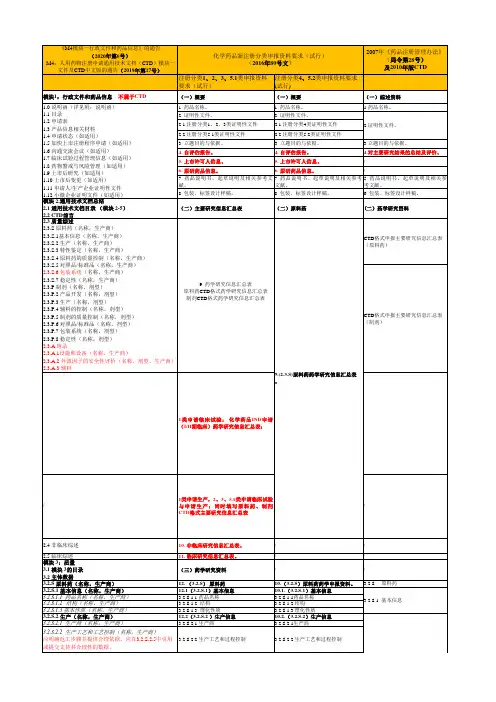
《M4模块一行政文件和药品信息》的通告(2020年第6号)M4:人用药物注册申请通用技术文档(CTD)模块一文件及CTD中文版的通告(2019年第17号)2007年《药品注册管理办法》(局令第28号)及2010年版CTD注册分类1、2、3、5.1类申报资料要求(试行)注册分类4、5.2类申报资料要求(试行)模块1:行政文件和药品信息不属于CTD(一)概要(一)概要(一)综述资料1. 药品名称。
1. 药品名称。
1.药品名称。
2. 证明性文件。
2. 证明性文件。
2.1 注册分类1、2、3类证明性文件 2.1 注册分类4类证明性文件2.2 注册分类5.1类证明性文件 2.2 注册分类5.2类证明性文件3. 立题目的与依据。
3. 立题目的与依据。
3.立题目的与依据。
4. 自评估报告。
4. 自评估报告。
4.对主要研究结果的总结及评价。
5. 上市许可人信息。
5. 上市许可人信息。
/6. 原研药品信息。
6. 原研药品信息。
/7.药品说明书、起草说明及相关参考文献。
7.药品说明书、起草说明及相关参考文献。
5.药品说明书、起草说明及相关参考文献。
8. 包装、标签设计样稿。
8. 包装、标签设计样稿。
6. 包装、标签设计样稿。
模块 2.通用技术文档总结2.1 通用技术文档目录(模块 2-5)2.2 CTD前言(二)主要研究信息汇总表(二)原料药(二)药学研究资料CTD格式申报主要研究信息汇总表(原料药)CTD格式申报主要研究信息汇总表(制剂)1类申请临床试验:化学药品IND申请(I/II期临床)药学研究信息汇总表;/1类申请生产,2、3、5.1类申请临床试验与申请生产:同时填写原料药、制剂CTD格式主要研究信息汇总表/2.4 非临床综述10. 非临床研究信息汇总表。
//2.5 临床综述11. 临床研究信息汇总表。
//模块 3:质量3.1 模块 3的目录3.2 主体数据(三)药学研究资料//3.2.S 原料药(名称,生产商)12. (3.2.S)原料药10.(3.2.S)原料药药学申报资料。

化药申报资料模版
化药申报资料模版通常包括以下几个部分:
1. 封面:封面应包括药品名称、申请企业信息、申请事项等内容。
2. 目录:列出资料各部分的标题,便于查找。
3. 证明文件:包括药品质量标准、药品生产工艺、药品注册申请表等证明文件的复印件或原件。
4. 药学研究资料:包括药品的理化性质、药效学研究、药代动力学研究、毒理学研究等方面的资料。
5. 临床研究资料:包括临床试验方案、临床试验报告、不良反应报告等方面的资料。
6. 生产工艺和检验方法研究资料:包括生产工艺流程图、工艺操作要点、质量控制标准等方面的资料。
7. 药品说明书和标签:包括药品说明书、标签和包装材料的复印件或原件。
8. 其他资料:根据具体申请事项,可能需要提供其他相关资料,如药品广告审查表等。
需要注意的是,不同国家和地区的化药申报资料要求可能有所不同,具体要求可以参考当地药品监管部门的相关规定。


附件化学药品新注册分类申报资料要求〔试行〕第一局部注册分类1、2、3、5.1类申报资料要求〔试行〕一、申报资料工程〔一〕概要1.药品名称。
2.证明性文件。
2.1注册分类1、2、3类证明性文件2.2注册分类5.1类证明性文件3.立题目的与依据。
4.自评估报告。
5.上市许可人信息。
6.原研药品信息。
7.药品说明书、起草说明及相关参考文献。
8. 包装、标签设计样稿。
〔二〕主要研究信息汇总表9. 药学研究信息汇总表。
10. 非临床研究信息汇总表。
11. 临床研究信息汇总表。
〔三〕药学研究资料12. 〔3.2.S〕原料药〔注:括号内为CTD格式的编号,以下同〕。
12.1〔3.2.S.1〕根本信息12.2〔3.2.S.2 〕生产信息12.3〔3.2.S.3 〕特性鉴定12.4〔3.2.S.4〕原料药的质量控制12.5〔3.2.S.5〕对照品12.6〔3.2.S.6〕包装材料和容器12.7〔3.2.S.7〕稳定性13. 〔3.2.P〕制剂。
13.1〔3.2.P.1〕剂型及产品组成13.2〔3.2.P.2〕产品开发13.3〔3.2.P.3〕生产13.4〔3.2.P.4〕原辅料的控制13.5〔3.2.P.5〕制剂的质量控制13.6〔3.2.P.6〕对照品13.7〔3.2.P.7〕稳定性〔四〕非临床研究资料14.非临床研究资料综述。
15.主要药效学试验资料及文献资料。
16.平安药理学的试验资料及文献资料。
17.单次给药毒性试验资料及文献资料。
18.重复给药毒性试验资料及文献资料。
19.遗传毒性试验资料及文献资料。
20.生殖毒性试验资料及文献资料。
21.致癌试验资料及文献资料。
22.依赖性试验资料及文献资料。
23.过敏性〔局部、全身和光敏毒性〕、溶血性和局部〔血管、皮肤、粘膜、肌肉等〕刺激性等特殊平安性试验资料及文献资料。
24.其他平安性试验资料及文献资料。
25.非临床药代动力学试验资料及文献资料。
26.复方制剂中多种成分药效、毒性、药代动力学相互影响的试验资料及文献资料。
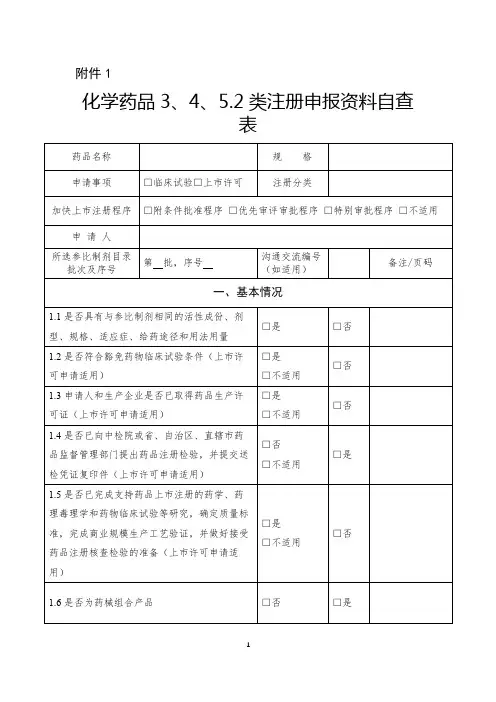

化学药品注册分类及申报资料要求(试行)第一部分注册分类1、2、3、5.1类申报资料要求(试行)一、申报资料项目(一)概要1.药品名称。
2.证明性文件。
2.1注册分类1、2、3类证明性文件2.2注册分类5.1类证明性文件3.立题目的与依据。
4.自评估报告。
5.上市许可人信息。
6.原研药品信息。
7.药品说明书、起草说明及相关参考文献。
8. 包装、标签设计样稿。
(二)主要研究信息汇总表9. 药学研究信息汇总表。
10. 非临床研究信息汇总表。
11. 临床研究信息汇总表。
(三)药学研究资料12. (3.2.S)原料药(注:括号内为CTD格式的编号,以下同)。
12.1(3.2.S.1)基本信息12.2(3.2.S.2 )生产信息12.3(3.2.S.3 )特性鉴定12.4(3.2.S.4)原料药的质量控制12.5(3.2.S.5)对照品12.6(3.2.S.6)包装材料和容器12.7(3.2.S.7)稳定性13. (3.2.P)制剂。
13.1(3.2.P.1)剂型及产品组成13.2(3.2.P.2)产品开发13.3(3.2.P.3)生产13.4(3.2.P.4)原辅料的控制13.5(3.2.P.5)制剂的质量控制13.6(3.2.P.6)对照品13.7(3.2.P.7)稳定性(四)非临床研究资料14.非临床研究资料综述。
15.主要药效学试验资料及文献资料。
16.安全药理学的试验资料及文献资料。
17.单次给药毒性试验资料及文献资料。
18.重复给药毒性试验资料及文献资料。
19.遗传毒性试验资料及文献资料。
20.生殖毒性试验资料及文献资料。
21.致癌试验资料及文献资料。
22.依赖性试验资料及文献资料。
23.过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等特殊安全性试验资料及文献资料。
24.其他安全性试验资料及文献资料。
25.非临床药代动力学试验资料及文献资料。
26.复方制剂中多种成分药效、毒性、药代动力学相互影响的试验资料及文献资料。

新注册分类 3 类与 4 类原料药申报资料要求比较国家局近日公布了《化学药品注册分类改革工作方案(征求意见稿)》,其中药品注册分类大家之前均已有了解,此次征求意见稿并无大的变化。
不过有两点内容应引起关注,一是监测期的变化,二是该方案中对于3 类、4类申报资料的要求的区别。
下面就3类与4类原料药申报资料的区别进行汇总如下:
(1)3类申报资料要求与387号文一致,没有变化。
而 4 类申报资料的要求应该是新撰写修订的,其中的许多要求更加具体、明确,便于研究人员理解操作,支持!
(2)4 类申报资料要求中多次提到了参考ICH相关指导原则,说明国内的研发与审评更加的与国际接轨了,研发人员更应该好好的研读ICH 的相关指导原则。
(3)4 类申报资料要求中多次提到了杂质研究,可见目前仿制药(特别是原料药)杂质研究仍然是高度关注的重点。
(4)4 类申报资料要求中明确了一些概念,并且与目前国内的核查要求衔接更加紧密,例如关键起始物料的供应商审计,应引起大家的重视。
(5)4 类中明确要求申报时需要提交12个月的长期稳定性试验数据,如此一来,国内仿制药研发的进度又会减慢了。
是一致的,且杂质含量不高于原研厂近效期的产。
新注册分类 3 类与 4 类原料药申报资料要求比较国家局近日公布了《化学药品注册分类改革工作方案(征求意见稿)》,其中药品注册分类大家之前均已有了解,此次征求意见稿并无大的变化。
不过有两点内容应引起关注,一是监测期的变化,二是该方案中对于3 类、4类申报资料的要求的区别。
下面就3类与4类原料药申报资料的区别进行汇总如下:
(1)3类申报资料要求与387号文一致,没有变化。
而 4 类申报资料的要求应该是新撰写修订的,其中的许多要求更加具体、明确,便于研究人员理解操作,支持!
(2)4 类申报资料要求中多次提到了参考ICH相关指导原则,说明国内的研发与审评更加的与国际接轨了,研发人员更应该好好的研读ICH 的相关指导原则。
(3)4 类申报资料要求中多次提到了杂质研究,可见目前仿制药(特别是原料药)杂质研究仍然是高度关注的重点。
(4)4 类申报资料要求中明确了一些概念,并且与目前国内的核查要求衔接更加紧密,例如关键起始物料的供应商审计,应引起大家的重视。
(5)4 类中明确要求申报时需要提交12个月的长期稳定性试验数据,如此一来,国内仿制药研发的进度又会减慢了。
是一致的,且杂质含量不高于原研厂近效期的产。
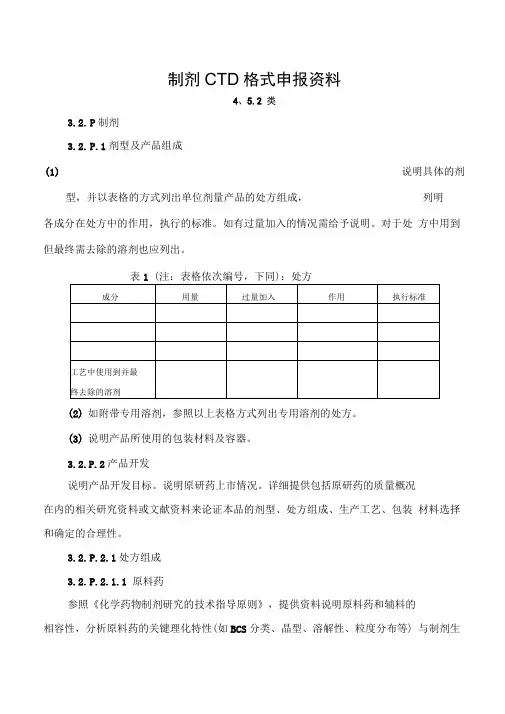
制剂CTD格式申报资料4、5.2 类3.2.P制剂3.2.P.1剂型及产品组成(1)说明具体的剂型,并以表格的方式列出单位剂量产品的处方组成,列明各成分在处方中的作用,执行的标准。
如有过量加入的情况需给予说明。
对于处方中用到但最终需去除的溶剂也应列出。
表1 (注:表格依次编号,下同):处方(2)如附带专用溶剂,参照以上表格方式列出专用溶剂的处方。
(3)说明产品所使用的包装材料及容器。
3.2.P.2产品开发说明产品开发目标。
说明原研药上市情况。
详细提供包括原研药的质量概况在内的相关研究资料或文献资料来论证本品的剂型、处方组成、生产工艺、包装材料选择和确定的合理性。
3.2.P.2.1处方组成3.2.P.2.1.1 原料药参照《化学药物制剂研究的技术指导原则》,提供资料说明原料药和辅料的相容性,分析原料药的关键理化特性(如BCS分类、晶型、溶解性、粒度分布等) 与制剂生产及制剂性能的相关性,并提供相关的研究资料与文献。
3.2.P.2.1.2 辅料说明辅料是否适合所用的给药途径结合辅料在处方中的作用分析辅料的哪些性质会影响制剂特性,提供相关的研究资料与文献。
3.2.P.2.2 制剂研究3.2.P.2.2.1处方开发过程参照《化学药物制剂研究的技术指导原则》,提供处方的研究开发过程和确定依据,包括文献信息(如对照药品的处方信息)、研究信息(包括处方设计,处方筛选和优化、处方确定等研究内容)、辅料种类和用量选择的依据、分析辅料用量是否在常规用量范围内,以及自制样品与原研药的质量特性对比研究结果(需说明原研药的来源、批次和有效期,自研样品批次,对比项目、采用方法),并重点说明在药品开发阶段中处方组成的主要变更、原因以及支持变化的验证研究。
如生产中存在过量投料的问题,应提供过量投料的必要性和合理性的相关研究资料。
3.2.P.2.2.2制剂相关特性对与制剂性能相关的理化性质,如pH、离子强度、溶出度、再分散性、复溶、粒径分布、聚合、多晶型、流变学等进行分析。
制剂CTD 格式申报资料4、5.2类3.2.P 制剂3.2.P.1 剂型及产品组成(1)说明具体的剂型,并以表格的方式列出单位剂量产品的处方组成,列明各成分在处方中的作用,执行的标准。
如有过量加入的情况需给予说明。
对于处方中用到但最终需去除的溶剂也应列出。
表1 (注:表格依次编号,下同):处方2)如附带专用溶剂,参照以上表格方式列出专用溶剂的处方。
3)说明产品所使用的包装材料及容器。
3.2.P.2 产品开发说明产品开发目标。
说明原研药上市情况。
详细提供包括原研药的质量概况在内的相关研究资料或文献资料来论证本品的剂型、处方组成、生产工艺、包装材料选择和确定的合理性。
3.2. P.2.1处方组成3.2. P.2.1.1原料药参照《化学药物制剂研究的技术指导原则》,提供资料说明原料药和辅料的相容性,分析原料药的关键理化特性(如BCS分类、晶型、溶解性、粒度分布等)与制剂生产及制剂性能的相关性,并提供相关的研究资料与文献。
3.2. P.2.1.2辅料说明辅料是否适合所用的给药途径结合辅料在处方中的作用分析辅料的哪些性质会影响制剂特性,提供相关的研究资料与文献。
3.2. P.2.2 制剂研究3.2. P.2.2.1处方开发过程参照《化学药物制剂研究的技术指导原则》,提供处方的研究开发过程和确定依据,包括文献信息(如对照药品的处方信息)、研究信息(包括处方设计,处方筛选和优化、处方确定等研究内容)、辅料种类和用量选择的依据、分析辅料用量是否在常规用量范围内,以及自制样品与原研药的质量特性对比研究结果(需说明原研药的来源、批次和有效期,自研样品批次,对比项目、采用方法),并重点说明在药品开发阶段中处方组成的主要变更、原因以及支持变化的验证研究。
如生产中存在过量投料的问题,应提供过量投料的必要性和合理性的相关研究资料。
3.2. P.2.2.2制剂相关特性对与制剂性能相关的理化性质,如pH、离子强度、溶出度、再分散性、复溶、粒径分布、聚合、多晶型、流变学等进行分析。
附件化学药品注册分类及申报资料要求一、化学药品注册分类化学药品注册分类分为创新药、改良型新药、仿制药、境外已上市境内未上市化学药品,分为以下5个类别:1类:境内外均未上市的创新药。
指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。
2类:境内外均未上市的改良型新药。
指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。
2.1含有用拆分或者合成等方法制得的已知活性成份的光学异构体,或者对已知活性成份成酯,或者对已知活性成份成盐(包括含有氢键或配位键的盐),或者改变已知盐类活性成份的酸根、碱基或金属元素,或者形成其他非共价键衍生物(如络合物、螯合物或包合物),且具有明显临床优势的药品。
2.2含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径,且具有明显临床优势的药品。
2.3含有已知活性成份的新复方制剂,且具有明显临床优势。
2.4含有已知活性成份的新适应症的药品。
—1—3类:境内申请人仿制境外上市但境内未上市原研药品的药品。
该类药品应与参比制剂的质量和疗效一致。
4类:境内申请人仿制已在境内上市原研药品的药品。
该类药品应与参比制剂的质量和疗效一致。
5类:境外上市的药品申请在境内上市。
5.1境外上市的原研药品和改良型药品申请在境内上市。
改良型药品应具有明显临床优势。
5.2境外上市的仿制药申请在境内上市。
原研药品是指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。
参比制剂是指经国家药品监管部门评估确认的仿制药研制使用的对照药品。
参比制剂的遴选与公布按照国家药品监管部门相关规定执行。
二、相关注册管理要求(一)化学药品1类为创新药,应含有新的结构明确的、具有药理作用的化合物,且具有临床价值,不包括改良型新药中2.1类的药品。
含有新的结构明确的、具有药理作用的化合物的新复方制剂,应按照化学药品1类申报。
新注册分类3类与4类原料药申报资料要求比较
国家局近日公布了《化学药品注册分类改革工作方案(征求意见稿)》,其中药品注册分类大家之前均已有了解,此次征求意见稿并无大的变化。
不过有两点内容应引起关注,一是监测期的变化,二是该方案中对于3类、4类申报资料的要求的区别。
下面就3类与4类原料药申报资料的区别进行汇总如下:
(1)3类申报资料要求与387号文一致,没有变化。
而4类申报资料的要求应该是新撰写修订的,其中的许多要求更加具体、明确,便于研究人员理解操作,支持!
(2)4类申报资料要求中多次提到了参考ICH相关指导原则,说明国内的研发与审评更加的与国际接轨了,研发人员更应该好好的研读ICH的相关指导原则。
(3)4类申报资料要求中多次提到了杂质研究,可见目前仿制药(特别是原料药)杂质研究仍然是高度关注的重点。
(4)4类申报资料要求中明确了一些概念,并且与目前国内的核查要求衔接更加紧密,例如关键起始物料的供应商审计,应引起大家的重视。
(5)4类中明确要求申报时需要提交12个月的长期稳定性试验数据,如此一来,国内仿制药研发的进度又会减慢了。
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全。
附件2化学药品注册受理审查指南(第二部分注册分类4、5.2类)(试行)国家食品药品监督管理总局2017年11月日可编辑范本目录一、适用范围 (1)二、资料受理部门 (1)三、申报资料基本要求 (1)(一)申请表的整理 (1)(二)申报资料的整理 (2)四、申请表审查要点 (6)五、申报资料审查要点 (11)(一)申报资料要求 (11)(二)资料审查内容 (12)六、受理审查决定 (16)(一)受理 (16)(二)补正 (16)(三)不予受理 (17)七、其他 (17)八、受理流程图 (17)附件:1.申报资料袋封面格式 (19)2.申报资料项目封面格式 (21)3.申报资料项目目录 (23)4.化学药品4、5.2类注册申报资料自查表 (24)化学药品注册受理审查指南(试行)第二部分注册分类4、5.2类一、适用范围化学药品注册分类4类仿制药申请;化学药品注册分类5.2类临床试验/上市申请。
二、资料受理部门由国家食品药品监督管理总局药品审评中心受理。
三、申报资料基本要求(一)申请表的整理1.种类与份数要求药品注册申请表、申报资料情况自查表各四份,一份为原件;药品研制情况申报表(如适用)、药品注册生产现场检查申请表(如适用)各四份,三份为原件。
2.依据《关于启用新版药品注册申请表报盘程序的公告》,申请表的填报须采用国家食品药品监督管理总局统一发布的填报软件,提交由新版《药品注册申请表报盘程序》生成的电子及纸质文件。
(确认所用版本为最新版[以最新发布的公告为准],所生成的电子文件的格式应为RVT文件。
各页的数据核对码必须一致,并须与提交的电子申请表一致,申请表及自查表各页边缘应加盖所有申请人或注册代理机构骑缝章。
)3.填写应当准确、完整、规范,不得手写或涂改,并应符合可编辑范本填表说明的要求。
(二)申报资料的整理1.数量与装袋方式2套完整申请资料(至少1套为原件)+1套综述资料(含概要及研究汇总表)复印件,每套装入相应的申请表。