2009 Elsevier Colloids and Surfaces A Physicochemical and Engineering Aspects
- 格式:pdf
- 大小:1.28 MB
- 文档页数:9

elsevier目录[隐藏]【爱思唯尔公司】【爱思唯尔公司部门介绍】【爱思唯尔公司发展里程碑】【Elsev ier数据库】爱思唯尔企业标志[编辑本段]【爱思唯尔公司】Our mission:Elsevi er is an integral p artn er with th e scien tifi c,techni cal and h ealth co mmuni ties,delivering superior inf ormation produ cts and servi ces that foster co mmuni cation,build insigh ts,and enabl e indi vidual and collecti ve advan cemen t in sci enti fic research and health car e.Elsevi er.Building insigh ts.Br eaking bound aries.爱思唯尔致力于为全球三千多万科学家、研究人员、学生、医学以及信息处理的专业人士提供一流的信息产品和革新性的工具。
我们很荣幸能在全球科技和医学学术团体中扮演一个不可或缺的角色并为这些领域的发展尽绵薄之力,帮助科研人员和专业人士提高生产力和效率,同时不断投入并努力创新来更好地满足全球学术社区的需要。
Els ev ier公司沿用了Elzev ir 书屋的名字,并将Elzev ir 改为更为现代的书写方式Els ev ier。
数百年沧桑,Elsev ier 已从一家小小的致力于传播经典学术的荷兰书店发展为一个向全球科技和医学学术群体提供超过20,000本的刊物和图书的国际化多媒体出版集团。
公司标志:爱思唯尔公司的标志为一个长者手执缠绕于一棵大树的藤条。
其中长者象征广大的科技工作者,大树象征已经获得的科学知识,而藤条则象征科学知识与科技工作者之间的联系。
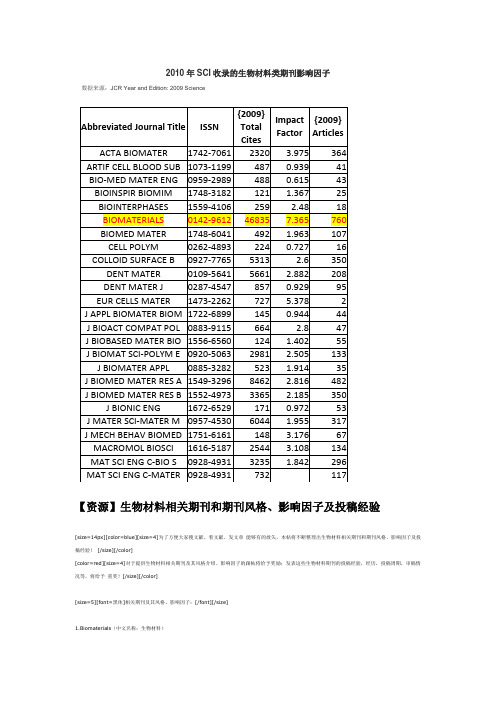
2010年SCI收录的生物材料类期刊影响因子数据来源:JCR Year and Edition: 2009 Science【资源】生物材料相关期刊和期刊风格、影响因子及投稿经验[size=14px][color=blue][size=4]为了方便大家搜文献、看文献、发文章能够有的放矢,本帖将不断整理出生物材料相关期刊和期刊风格、影响因子及投稿经验![/size][/color][color=red][size=4]对于提供生物材料相关期刊及其风格介绍、影响因子的跟帖将给予奖励;发表这些生物材料期刊的投稿经验,经历,投稿周期,审稿情况等,将给予重奖![/size][/color][size=5][font=黑体]相关期刊及其风格、影响因子:[/font][/size]1.Biomaterials(中文名称:生物材料)网址:[url=/]http://www.biomat [/url]07年IF:6.262生物材料最专业的两个期刊之一。
主要展示生物材料的应用以及相关医疗器械的研究成果。
发表关于临床应用材料中最重要问题的研究原著及权威性的综述。
文章涉及生物材料的基础科学和工程学等方面,包括力学、物理学、化学以及生物学特性,相关设计以及用这些材料制造的机械的产品特征和临床性能。
2.Journal of Biomedical Materials Research(中文名称:生物医学材料研究)网址:Part A[url=/journal/117935006/grouphome/home.html]/journal/1179 35006/grouphome/home.html[/url]Part B [url=/journal/117935007/grouphome/home.html]/journal/1179 35007/grouphome/home.html[/url]06年IF:2.497生物材料最专业的两个期刊之一,内容跟Biomaterials非常相似。


序号杂志全名中译名ISSN 2009版影响因子1 ADV ANCED MATERIALS 先进材料0935-9648 8.3792 PROGRESS IN SURFACE SCIENCE 表面科学进展0079-6816 7.9133 ANNUAL REVIEW OF MATERIALS RESEARCH 材料研究年度评论1531-7331 7.9114 INTERNA TIONAL MATERIALS REVIEWS 国际材料评论0950-6608 4.8575 CURRENT OPINION IN SOLID STATE & MATERIALS SCIENCE 固态和材料科学的动态1359-0286 4.0006 Langmuir 朗缪尔0743-7463 3.8987 Acta materialia 材料学报1359-6454 3.7608 BIOMETALS 生物金属0966-0844 3.1729 SCRIPTA MA TERIALIA 材料快报1359-6462 2.94910 COMPOSITES SCIENCE AND TECHNOLOGY 复合材料科学与技术0266-3538 2.90111 Nanoscale Research Letters 纳米研究快报1931-7573 2.89412 Science and Technology of Advanced Materials 先进材料科学技术1468-6996 2.59913 JOURNAL OF BIOMATERIALS SCIENCE-POL YMER EDITION 生物材料科学—聚合物版0920-5063 2.50514 COMPOSITES PART A-APPLIED SCIENCE AND MANUFACTURING 复合材料A应用科学与制备1359-835X 2.41015 Journal of Solid State Chemistry 固体化学0022-4596 2.34016 CORROSION SCIENCE 腐蚀科学0010-938X 2.31617 INTERMETALLICS 金属间化合物0966-9795 2.23118 Mechanics of Materials 材料力学0167-6636 2.20619 Solid State Ionics 固体离子0167-2738 2.16220 JOURNAL OF ALLOYS AND COMPOUNDS 合金和化合物杂志0925-8388 2.13521 JOURNAL OF THE EUROPEAN CERAMIC SOCIETY 欧洲陶瓷学会杂志0955-2219 2.09022 MA TERIALS CHEMISTRY AND PHYSICS 材料化学与物理0254-0584 2.01523 COMPOSITE STRUCTURES 复合材料结构0263-8223 2.00624 Materials Letters 材料快报0167-577X 1.94025 SYNTHETIC METALS 合成金属0379-6779 1.90126 MA TERIALS SCIENCE AND ENGINEERING A-STRUCTURAL MATERIALS PROPERTIES MICROST 材料科学和工程A—结构材料的性能、组织与加工0921-5093 1.90127 MA TERIALS RESEARCH BULLETIN 材料研究公告0025-5408 1.87928 SURFACE SCIENCE 表面科学0039-6028 1.79829 SURFACE & COA TINGS TECHNOLOGY 表面与涂层技术0257-8972 1.79330 Wear 磨损0043-1648 1.77131 ADV ANCED ENGINEERING MA TERIALS 先进工程材料1438-1656 1.76132 MA TERIALS SCIENCE AND ENGINEERING B-SOLID STA TE MATERIALS FOR ADV ANCED TECH 材料科学与工程B—先进技术用固体材料0921-5107 1.75633 INTERNA TIONAL JOURNAL OF REFRACTORY METALS & HARD MATERIALS 耐火金属和硬质材料国际杂志0263-4368 1.75034 SMART MATERIALS & STRUCTURES 智能材料与结构0964-1726 1.74935 THIN SOLID FILMS 固体薄膜0040-6090 1.72736 MATERIALS RESEARCH INNOV ATIONS 材料研究创新1432-8917 1.72337 COMPOSITES PART B-ENGINEERING 复合材料B工程1359-8368 1.70438 Solid State Sciences 固体科学1293-2558 1.67539 IEEE Transactions on Nanotechnology IEEE 纳米学报1536-125X 1.67140 JOURNAL OF MATERIALS RESEARCH 材料研究杂志0884-2914 1.66741 APPLIED SURFACE SCIENCE 应用表面科学0169-4332 1.61642 INTERNA TIONAL JOURNAL OF FATIGUE 疲劳国际杂志0142-1123 1.60243 METALLURGICAL AND MA TERIALS TRANSACTIONS A-PHYSICAL METALLURGY AND MATERIAL 冶金与材料会刊A——物理冶金和材料1073-5623 1.56444 Philosophical Magazine Letters 哲学杂志(包括材料)0950-0839 1.53045 COMPUTATIONAL MATERIALS SCIENCE 计算材料科学0927-0256 1.52246 MATERIALS & DESIGN 材料与设计0261-3069 1.51847 Current Nanoscience 当代纳米科学1573-4137 1.47248 JOURNAL OF MATERIALS SCIENCE 材料科学杂志0022-2461 1.47149 EUROPEAN PHYSICAL JOURNAL B 欧洲物理杂志 B 1434-6028 1.46650 JOURNAL OF V ACUUM SCIENCE & TECHNOLOGY B 真空科学与技术杂志B 1071-1023 1.46051 Journal of Nanoscience and Nanotechnology 纳米科学和纳米技术1533-4880 1.43552 JOURNAL OF MATERIALS PROCESSING TECHNOLOGY 材料加工技术杂志0924-0136 1.42053 MATERIALS CHARACTERIZATION 材料表征1044-5803 1.41654 JOURNAL OF SOL-GEL SCIENCE AND TECHNOLOGY 溶胶凝胶科学与技术杂志0928-0707 1.39355 JOURNAL OF V ACUUM SCIENCE & TECHNOLOGY A-V ACUUM SURFACES AND FILMS 真空科学与技术A真空表面和薄膜0734-2101 1.29756 Philosophical Magazine 哲学杂志1478-6435 1.27357 JOURNAL OF NON-CRYSTALLINE SOLIDS 非晶固体杂志0022-3093 1.25258 International Journal of Nanotechnology 纳米工程1475-7435 1.23459 PHYSICA STATUS SOLIDI A-APPLIED RESEARCH 固态物理A——应用研究1862-6300 1.22860 JOURNAL OF PHYSICS AND CHEMISTRY OF SOLIDS 固体物理与化学杂志0022-3697 1.18961 JOURNAL OF ADHESION SCIENCE AND TECHNOLOGY 粘着科学与技术杂志0169-4243 1.17562 PHYSICA STATUS SOLIDI B-BASIC RESEARCH 固态物理B—基础研究0370-1972 1.15063 Oxidation of Metals 材料氧化0030-770X 1.10864 METALS AND MATERIALS INTERNATIONAL 国际金属及材料1598-9623 1.09065 MODERN PHYSICS LETTERS A 现代物理快报A 0217-7323 1.07566 PHYSICA B 物理B 0921-4526 1.05667 NANO 纳米1793-2920 1.00868 IEEE TRANSACTIONS ON SEMICONDUCTOR MANUFACTURING IEEE半导体制造会刊0894-6507 1.00769 SURFACE AND INTERFACE ANAL YSIS 表面与界面分析0142-2421 0.99870 Vacuum 真空0042-207X 0.97571 MA TERIALS AND MANUFACTURING PROCESSES 材料与制造工艺1042-6914 0.96872 CORROSION 腐蚀0010-9312 0.95273 IEEE TRANSACTIONS ON COMPONENTS AND PACKAGING TECHNOLOGIES IEEE元件及封装技术会刊1521-3331 0.94474 PHASE TRANSITIONS 相变0141-1594 0.93575 APPLIED COMPOSITE MATERIALS 应用复合材料0929-189X 0.93576 METALLURGICAL AND MA TERIALS TRANSACTIONS B-PROCESSMETALLURGY AND MA TERIALS 冶金和材料会刊B—制备冶金和材料制备科学1073-5615 0.93277 Journal of Computational and Theoretical Nanoscience 计算与理论纳米科学1546-1955 0.89978 JOURNAL OF THE CERAMIC SOCIETY OF JAPAN 日本陶瓷学会杂志1882-0743 0.86279 International Journal of materials research 材料研究杂志1862-5282 0.86280 JOURNAL OF MATERIALS SCIENCE & TECHNOLOGY 材料科学与技术杂志1005-0302 0.82881 JOURNAL OF ENGINEERING MA TERIALS AND TECHNOLOGY-TRANSACTIONS OF THE ASME 工程材料与技术杂志—美国机械工程师学会会刊0094-4289 0.81582 JOURNAL OF COMPOSITE MA TERIALS 复合材料杂志0021-9983 0.80683 International Journal of Fracture 断裂学报0376-9429 0.80484 MATERIALS TRANSACTIONS 材料会刊1345-9678 0.79585 MATERIALS SCIENCE AND TECHNOLOGY 材料科学与技术0267-0836 0.79486 Journal of Porous Materials 多孔材料1380-2224 0.78887 BULLETIN OF MATERIALS SCIENCE 材料科学公告0250-4707 0.78388 RESEARCH IN NONDESTRUCTIVE EVALUATION 无损检测研究0934-9847 0.76089 EUROPEAN PHYSICAL JOURNAL-APPLIED PHYSICS 欧洲物理杂志—应用物理1286-0042 0.75690 MATERIALS AND STRUCTURES 材料与结构1359-5997 0.75391 SYNTHESE 合成0039-7857 0.72992 SCIENCE IN CHINA SERIES E-TECHNOLOGICAL SCIENCES 中国科学E技术科学1006-9321 0.68293 COMPOSITE INTERFACES 复合材料界面0927-6440 0.67094 Mechanics of Advanced Materials and Structures 先进材料结构和力学1537-6494 0.65895 RARE METALS 稀有金属1001-0521 0.60196 MATERIALS TECHNOLOGY 材料技术1066-7857 0.59797 JOURNAL OF MATERIALS ENGINEERING AND PERFORMANCE 材料工程与性能杂志1059-9495 0.59298 Journal of RARE EARTH 稀土学报1002-0721 0.57299 Reviews on Advanced Materials Science 先进材料科学评论1606-5131 0.558100 ADV ANCED COMPOSITE MA TERIALS 先进复合材料0924-3046 0.500101 ACTA METALL SIN 金属学报0412-1961 0.483102 TRANSACTIONS OF NONFERROUS METALS SOCIETY OF CHINA 中国有色金属学会会刊1003-6326 0.445103 Surface Engineering 表面工程0267-0844 0.432104 Journal of Phase Equilibria and Diffusion 相平衡与扩散1547-7037 0.415105 INTERNATIONAL JOURNAL OF MODERN PHYSICS B 现代物理国际杂志B 0217-9792 0.408106 INTERNATIONAL JOURNAL OF MATERIALS & PRODUCT TECHNOLOGY 材料与生产技术国际杂志0268-1900 0.384107 SURFACE REVIEW AND LETTERS 表面评论与快报0218-625X 0.366108 JOURNAL OF WUHAN UNIVERSITY OF TECHNOLOGY-MATERIALS SCIENCE EDITION 武汉理工大学学报-材料科学版1000-2413 0.352109 HIGH TEMPERATURE MATERIALS AND PROCESSES 高温材料和加工0334-6455 0.351110 ADV ANCED COMPOSITES LETTERS 先进复合材料快报0963-6935 0.311111 MA TERIALS EV ALUATION 材料评价0025-5327 0.288112 Solid State Technology 固体物理技术0038-111X 0.275113 AMERICAN CERAMIC SOCIETY BULLETIN 美国陶瓷学会公告0002-7812 0.268114 JOURNAL OF ADV ANCED MATERIALS 先进材料杂志1070-9789 0.245115 POWDER METALLURGY AND METAL CERAMICS 粉末冶金及金属陶瓷1068-1302 0.238116 MATERIALS SCIENCE 材料科学1068-820X 0.231 117 ADV ANCED MA TERIALS & PROCESSES 先进材料及工艺0882-7958 0.199118 RARE METAL MATERIALS AND ENGINEERING 稀有金属材料与工程1002-185X 0.161119 SCIENCE AND ENGINEERING OF COMPOSITE MA TERIALS 复合材料科学与工。

荷兰Elsevier公司旗下的英⽂全⽂数据库荷兰Elsevier公司旗下的英⽂全⽂数据库-----Science Direct,Wiley数据库――科学研究的必备之选。
现在好了,各⼤出版社均推出了在线投稿系统,这⼏年把⼏个⼤的投稿系统都试过了,总结⼀下谈谈对⼏个投稿系统的感受吧。
1)Nature系列期刊这个系列期刊在线投稿系统⽐较烦,我们当时光研究投稿须知就研究了⼀天,它对稿件要求很具体,也很苛刻,每个图都要独⽴作成单独的⽂件,如果⼀张图由⼏张图构成,还要说明这图是如何由这⼏部分构成的。
不过现在Nature系列期刊都可以先写⼀个⽐较详细的Coverletter过去,介绍⾃已的⼯作,然后主编决定是否让你投稿,这样可以省不少事,因为⼤部分主编看⼀眼就会拒掉,可能你花在投稿上的时间远⽐他看你⽂章的时间长得多。
2)Science系列同为顶级期刊,Science的在线投稿系统简单了许多,你可以将⽂章和Coverletter分别作成⼀个⽂件上传即可,不过基本上也是⼤部分很快被拒,偶们的⽂章在投稿后三天被拒。
3)美国化学会Journal of the American Chemical Society 《美国化学学会会志》ACS Nano 《ACS纳⽶》Analytical Chemistry * 《分析化学》ACS Chemical Biology 《ACS化学⽣物学》Biochemistry 《⽣物化学》Energy & Fuels 《能源和燃料》个⼈⼀直认为美国化学会的期刊是化学类杂志极有代表性的期刊,象JACS,NL都是学界顶尖杂志。
美国化学会在线投稿系统作得也⽐较完善,⼀般要求将⽂章与Coverletter还有Surpporting Information(如果有的话)分别作成单独的⽂件上传,即完成投稿,系统会⾃动为你转成PDF格式,当然你也可以直接上传PDF,但⼀般都还要求同时上传Word⽂件,总体⽽⾔,对美国化学会的在线投稿系统映像是很不错的。

向老外作者要文献的一个常用的模板Dear Professor ×××I am in ××× Institute of ×××, Chinese Academy of Sciences.I am writing to request your assistance. I search one of your papers:。
(你的文献题目)but I can not read full-text content, would you mind sending your papers by E-mail? Thank you for your assistance.Best wishes !(or best regards)×××这个暑假中了2篇SCI文章,影响因子都在IF=1.5-2.0之间。
其实,在此之前,本人已经发表了若干SCI,而且已经是两个期刊的Reviewer。
但尽管如此,随着文章积累越多,对SCI写作的认识也有所熟悉和深入。
下面谈谈一些体会,与大家分享。
第一篇:去年12月份投稿,7月份返回意见。
结论是:“I am pl eased to inform you that your paper has been accepted for publication provided that you amend it according to the concerns raised in the review report given below.”实际上,这个结论已经非常好了。
我看了以下审稿意见,然后就逐条的进行了Response。
其中Response letter的格式我是参考了我审稿过的一篇德国学者的回复模式(我认为非常好)。
但是,在审稿意见中,有一条意见要我对实验过程做一描述。

Colloids and Surfaces B:Biointerfaces 65(2008)239–246Contents lists available at ScienceDirectColloids and Surfaces B:Biointerfacesj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /c o l s u r fbPhysico-chemical properties and cytotoxicity assessment of PEG-modified liposomes containing human hemoglobinVal ´erie Centis a ,Patrick Vermette a ,b ,∗aLaboratoire de Bioing´e nierie et de Biophysique de l’Universit´e de Sherbrooke,Department of Chemical Engineering,Universit´e de Sherbrooke,2500,boul.de l’Universit´e ,Sherbrooke (QC),Canada J1K 2R1bResearch Centre on Aging,Institut universitaire de g´e riatrie de Sherbrooke,1036,rue Belv´e d`e re Sud,Sherbrooke (QC),Canada J1H 4C4a r t i c l e i n f o Article history:Received 5December 2007Received in revised form 14April 2008Accepted 15April 2008Available online 24April 2008Keywords:PEGylated liposomes encapsulating hemoglobin Oxygen carriers Cytotoxicity HUVECCharacterizationTransmission electron microscopy Zeta potentialParticle size distributiona b s t r a c tPEGylated liposomes encapsulating human hemoglobin as oxygen carriers were prepared frompurified carbonylhemoglobin (HbCO)solution and a lipid mixture composed of 1,2-dipalmitoyl-sn -glycero-3-phosphatidylcholine (DPPC),cholesterol,1,2-dimyristoyl-sn -glycero-3-phosphoethanolamine-N -[poly(ethylene glycol)2000](DMPE-PEG 2000)and palmitic acid.Hemoglobin was extracted and purified from human blood samples.SDS-PAGE was used to assess its purity.Diameter of liposomes containing hemoglobin was controlled to approximately 200nm using extrusion as measured by dynamic light scattering and transmission electron microscopy.Liposome size distributions were shown to remain unimodal over 14days,even at different storage temperatures.Zeta potential measurements revealed that liposome containing hemoglobin have a net surface charge of −7.16±0.33mV.Also,hemoglobin encapsulated in liposomes was able to perform several cycles of oxygen loading and unloading using oxygen (O 2)and carbon monoxide (CO).The hemoglobin vesicle dispersion showed some toxicity as revealed by three in vitro assays in which endothelial cell (HUVECs)monolayers were exposed to these dispersions.Cytotoxicity was function of the liposome concentration in the culture medium.©2008Elsevier B.V.All rights reserved.1.IntroductionSince Rabiner et al.in 1967and Savitsky et al.in 1978[1,2]stud-ied stroma-free hemoglobin solution as a possible substitute for human blood,research on the development of universal,safe and effective oxygen carriers has been an ongoing subject.Such a solu-tion,despite its purpose to be used in blood replacement therapy,could also be used to supply oxygen in high-density cell culture sys-tems,where oxygen is often the limiting factor.This is particularly flagrant in tissue engineering applications [3–7].Many perfluorocarbon emulsions and hemoglobin-based prepa-rations have been considered as oxygen carriers,with their loads of advantages and disadvantages.For a complete review on the dif-ferent types of oxygen carriers,their design and characterization,the reader is referred to Riess [8].Liposomes and biodegradable polymer capsules have been examined as confinement vehicles for tetrameric hemoglobin [9–12].Liposomes are double-layered phospholipid vesicles able∗Corresponding author.Tel.:+18198218000x62826;fax:+18198217955.E-mail address:Patrick.Vermette@USherbrooke.ca (P.Vermette).to encapsulate different drugs or molecules [13].They have been thoroughly studied for the past 40years and the reader is directed to the work of Lasic and colleagues for more information [13–15].Surface modification with PEG was reported to improve the cir-culation of liposomes in circulation [8,16].PEG is a biologically inert polymer extensively used in drug delivery to create,once conjugated,a steric barrier and/or a water structure around lipo-somes,with the aim to protect liposomes from plasma proteins in the body [17,18].The steric barrier created by PEG conjugation also prevents liposome fusion and aggregation and thus,stabilizes the dispersions during storage [17,18].It was shown by Sakai et al.[17]that PEGylated liposomes encapsulating hemoglobin stored in a deoxygenated state at 4◦C and 23◦C were stable for 1year,while dispersions stored at 40◦C were stable for 6months before liposome aggregation and hemoglobin leakage were observed [17].To be used in cell cultures,liposome dispersions should have limited cell toxicity and their formulations should be stable throughout the duration of the culture.Also,the liposome prepa-ration should be able to increase the oxygen concentration in the cultures and therefore,have a positive impact on the cell growth.Cytotoxicity of PEGylated liposomes encapsulating hemoglobin was assayed in different studies.For example,hemoglobin vesicles0927-7765/$–see front matter ©2008Elsevier B.V.All rights reserved.doi:10.1016/j.colsurfb.2008.04.009240V.Centis,P.Vermette/Colloids and Surfaces B:Biointerfaces65(2008)239–246were tested on human cord blood hematopoietic progenitor cells [19].Mild toxicity was observed on short-term exposures,while inhibition of proliferation was monitored at longer times[19].Also, the effect of liposome encapsulating hemoglobin(LEH)on human platelet function in plasma was studied.Wakamoto et al.observed neither aggregation,activation,nor adverse effects when human platelets were exposed to liposome dispersions[20].Similarly,the exposure of rat erythrocytes to PEG-LEH did not affect the struc-tural integrity of the blood structures[21].No signs of aggregation and deformation of the erythrocytes were observed,suggesting that liposomes exhibited low,if any toxicity[21].Another study examined the cytokine-induced adhesiveness of monocytic cells to HUVECs[22].The data suggested that,in this specific model,LEH did not induce leukocyte adhesion and could have a beneficial effect in preventing leukocyte adhesion to vascular endothelium caused by inflammatory cytokines[22].To our knowledge,in vitro long-term cytotoxicity of LEH formulations towards human cells have not been fully addressed and such testing is afirst and manda-tory step to investigate the applicability of these LEH in tissue engineering.Therefore,this work examines the preparation and characteri-zation of liposomes encapsulating human hemoglobin as potential oxygen carriers for tissue engineering purposes and presents a char-acterization of the LEH dispersions.To assess the feasibility of using LEH as oxygen transporters,size distribution,zeta potential,mor-phology and shape,and cytotoxicity of these LEH dispersions were examined.To our knowledge,no similar study has been reported.2.Materials and methods2.1.Materials1,2-Dipalmitoyl-sn-glycero-3-phosphatidylcholine(DPPC,cat. #PCS-020)and1,2-dimyristoyl-sn-glycero-3-phosphoethanol-amine-N-[poly(ethylene glycol)2000](DMPE-PEG2000,cat.#PPE-010)were purchased from Northern Lipids(Vancouver,BC, Canada).Cholesterol(Chol,cat.#110525)was obtained from Avanti Polar Lipids(Alabaster,AL,USA).Palmitic acid(PA,cat.#P-0500),Medium199(M-199,cat.M-5017),Heparin(cat.#H-1027), Hank’s balances salt solution(HBSS,cat.#H-6648),fetal bovine serum(FBS,cat.#F-1051),endothelial cell growth supplement (ECGS,cat.#E-2759),pyridoxal5 -phosphate(PLP,cat.#P-3657) and dl-homocysteine(Hcy,cat.#H-4628)were purchased from Sigma–Aldrich(Oakville,ON,Canada).Chloroform,sodium dithionite,and dichloromethane used for hemoglobin purification and liposome preparation were of analyt-ical grade and were obtained from Fisher Chemicals.All the water used in these experiments was taken from a Milli-Q Gradient(Milli-pore,Billerca,MA,USA)ultra-pure deionization water system with a resistivity of18.2M cm.CellTiter96®AQueous One Solution Cell Proliferation Assay Kit(cat.#G3580)and CytoTox96®Non-Radioactive Cytotoxicity Assay(Lactate deshydrogenase,cat.#G1780)were obtained from Promega(Madison,WI,USA).CyQuant®NF Cell Proliferation Assay Kit(cat.#C35006)was purchased from Invitrogen(Burlington,ON, Canada).2.2.Hemoglobin purificationHemoglobin was obtained from human whole blood of healthy volunteers.All procedures were approved by the Ethics Committee of the Research Centre on Aging(Sherbrooke,Qc).After the subjects were thoroughly informed about the nature and goal of the study, they provided written consent.The protein was purified accord-ing to an adapted method previously published[23].In summary: (1)hemoglobin was stabilized by CO complexion(HbCO)(60min at room temperature);(2)the removal of membranes and stroma was done by solvent treatment(20%(v/v)CH2Cl2,shaken for3min with subsequent centrifugation at1900×g for15min);(3)traces of dichloromethane were removed by slowly heating the sample with aflow of inert gas(Ar)(heating from40◦C to60◦C,120min);(4)virus inactivation was done by heating the hemoglobin solu-tion(60◦C,overnight,with magnetic stirring agitation);(5)dialysis (MW cutoff of25kDa,against ultra-pure water,for24h,with mag-netic stirring agitation)was used to remove small molecules;(6) ultra-filtration(MW cutoff of30kDa,Millipore,USA)was used to filter out molecules with MW<30kDa.Solutions containing puri-fied HbCO were aliquoted and preserved in liquid nitrogen with PLP(18mM)and Hcy(15mM).SDS-PAGE was used to assess the presence of hemoglobin.2.2.1.SDS-PAGEThe purification of Hb was determined via SDS-PAGE using a FisherBiotech system(Fisher Scientific,cat.#FB200).All the sam-ples were prepared in a Laemmli buffer(Bio-rad,cat.#161–0158) with a4%acrylamide stacking gel and a15%acrylamide resolving gel(Bio-rad,cat.#161–0158).Human hemoglobin is known to have a MW of approximately 64kDa[8,24].The hemoglobin samples were run along with a pre-stained broad-range molecular weight marker(Bio-rad,cat. #161–0318),which consisted of proteins having molecular weights ranging between6.9and210kDa.10L of hemoglobin samples (2g/mL)and7L of the molecular weight marker(dilution ratio of1:75(v/v)i.e.,1part of the marker in75parts of a buffer)were deposited in each well.A purified commercial hemoglobin sam-ple(Sigma,cat.#H7379)was used as a control and was run at a concentration of0.1g/mL.All samples were run at115V until the migration front reached the bottom of the gel(±120min).The gels were then stained using Silver SNAP Stain kit II(Fisher Scientific, cat.#24612)to determine band intensities.2.3.Liposome preparationLiposomes were prepared in a slightly different way than the method previously reported[23].Briefly,the mixture of phos-pholipids DPPC,Chol,PA and DMPE-PEG2000in a molar ratio of 5/5/1/(0.3%[mole/total moles of lipids])was dissolved in chloro-form.The organic solvent was then evaporated using a rotary evaporator(B¨uchi,Switzerland)under reduced pressure to remove all traces of solvent.The resulting thinfilm was then hydrated with a NaOH solution(0.9mM)overnight to allow thefilm to dissolve to yield a lipid concentration of50mg/mL.The solution was then diluted with pure water to obtain a concentration of25mg/mL of lipids.Then,ten cycles of freeze-thawing(−196◦C and40◦C)were performed.The solution was then frozen for10min in liquid nitro-gen and lyophilized for48h.After dispersing the lipid mixture into a hemoglobin solution(4g/mL)for2h,the resulting multilamel-lar vesicles were extruded with the LiposoFast-Basic(Avestin Inc, Ottawa,ON,Canada)using11passes at54◦C through polycarbon-ate membranes with pore sizes of200nm.The resulting solution was then ultra-centrifuged at50,000×g for30min(Optima TLX, Beckman Coulter,Mississauga,On)to recover and eliminate the top layer containing non-encapsulated Hb.2.4.Liposome characterization2.4.1.Particle size distributionThe intensity mean diameter of the liposomes and the polydis-persity index(PI)of the distribution were determined by dynamicV.Centis,P.Vermette/Colloids and Surfaces B:Biointerfaces65(2008)239–246241light scattering(DLS)using a Zetasizer Nanoseries(Malvern Instru-ments,UK).Samples of liposomes stored at4◦C,22◦C and37◦C were analyzed.The refractive index and viscosity of pure water were used as calculation parameters and each sample was mea-sured3times for25runs using the unimodal model for size distribution.All samples were diluted to an appropriate counting rate prior to analysis.2.4.2.Zeta potentialThe zeta potential was measured by electrophoresis at25◦C (Zetasizer Nanoseries,Malvern Instruments,UK).The surface charge of the particles was obtained by measuring the velocity of the dispersion in an electricalfield.Samples were diluted and placed in an electrophoretic cell where a potential of±60mV was established.Actual values were calculated from the mean elec-trophoretic mobility using Smoluchowski’s equation.The viscosity and dielectric constant of pure water were used as calculation parameters and each sample was measured in quadruplicate.All samples were diluted to an appropriate counting rate prior to anal-ysis.2.4.3.Influence of pH and osmolaritySlight changes in pH and osmolarity can have deleterious effects on cells in culture.To ensure that seeded cells would not respond to a change in pH and osmolarity due to the addition of liposomes in culture media,these parameters were moni-tored over24h(6,12,and24h).HUVECs were seeded onto 24-well plates(6000cells/well)(Corning,cat.#3527)and let to adhere in normal incubator conditions.The next day,HUVECs were either exposed to:(1)LEH(2mg/mL),(2)“Empty”lipo-somes i.e.,liposomes with no hemoglobin(2mg/mL),or(3)M-199 media.After6,12and24h,250L were pipetted and used for osmolarity analyses using a pre-calibrated osmometer(Advanced Instruments,model3250,Norwood,MS,USA).The rest of the solution was used for pH monitoring(Orion Research,model 710A,Lausanne,Switzerland).Measurements were done in triplicate.2.5.Functionality of the LEH as oxygen carriersHemoglobin possesses characteristic bands throughout the vis-ible spectra.Q-band(around500–600nm)and Soret band(in the blue region,around400nm)are typical regions that provide infor-mation about the hemoglobin state.Therefore,functionality of hemoglobin entrapped in liposomes and its ability to shift from its oxygen-loaded position to unloaded position was assayed by spectrometry.LEH dispersions(2mg/mL,using HBSS buffer)were exposed to pure oxygen and halogen light(90min,400W)[25]and sub-sequently compared to carbon monoxide(CO)exposure(15min, in the dark).To obtain deoxygenated spectra of hemoglobin,solu-tions were equilibrated with inert gas(Ar)to remove most of the oxygen before treatment with dithionite[26].Wavelengths shifts of the Q-band and Soret band under these conditions were mea-sured using a micro-plate reader(Bio-Tek Instruments,Winooski, VT,USA).2.6.Transmission electron microscopy(TEM)analysesThe samples were diluted appropriately and were negatively stained with uranyl acetate and allowed to air-dry directly on TEM grids.Observations were made using an H-7500transmission elec-tron microscope(HITASHI,Pleasanton,CA,USA)at a voltage of 60kV.2.7.HUVEC cultureHuman Umbilical Vein Endothelial Cells(HUVEC,PromoCell, Heidelberg,Germany)were cultured in M-199supplemented with10%(v/v)de-complemented FBS.ECGS(20g/mL),heparin (90mg/L),and antibiotics(100U/mL penicillin G and100g/mL streptomycin)were also added.Culture media was replenished three times a week.All cells were maintained at37◦C in an incu-bator with a humidified atmosphere containing5%CO2.Cells of passages4and5were used throughout this study.2.8.Cytotoxicity of LEHFollowing cell exposure to LEH,cell proliferation assays were performed to evaluate the cytotoxicity of the LEH formulations.For this purpose,HUVECs were incubated with three concentrations of LEH dispersions(5,2and1mg/mL)for6,12,and24h.The M-199culture medium concentration was adjusted to compensate for the dilution caused by the addition of liposomes.Empty liposomes (5mg/mL)were used as controls.Three assays were performed to evaluate the cytotoxicity of the LEH dispersions.Different assays were used since it was reported that cellular metabolic processes vary greatly over time and that methodologies that rely only on measurements of ATP content can cause some problems in further cytotoxicity interpretation[27].LEH cytotoxicity wasfirst examined using the MTS assay,a colorimetric method optimized for adherent cells.Viable cells enzymatically reduce the colorless tetrazolium salt MTS to inten-sively colored MTS-formazan.Briefly,4000cells/well were seeded inflat bottom96-well plates(Corning Incorporated,Corning,NY, USA)and incubated for24h.After removing the culture medium, 100L of LEH dispersions were applied,and the plates were incu-bated at37◦C for a period of4h.The absorbance was read at490nm on a micro-plate reader(Bio-Tek Instruments,Winooski,VT,USA). Values were corrected for background absorbance.Secondly,cell number was assessed using the CyQuant®Cell Proliferation Assay Kit,a highly sensitive,fluorescence-based micro-plate assay[27].The CyQuant®assay measures the ability of CyQuant®dye to bind to the cellular nucleic acids of viable cells. Measurements of cellular proliferation provide a general measure of toxicity.Cell cultures were seeded at103cells/well in duplicate inflat bottom96-well plates.After exposure of the liposomes to adherent cells,cells were rinsed with HBSS buffer to remove dead cells no longer adhering to the plate,lysed,and the DNA was stained using the CyQuant®fluorescent dye solution as recommended by the manufacturer.Plates were incubated at37◦C for60min.Flu-orescence was measured using afluorescence micro-plate reader (Bio-Tek Instruments,Winooski,VT,USA).The excitation maximum was480nm and the emission maximum was530nm.Values were corrected for background absorbance.Finally,quantification of the release of the cytoplasmic enzyme, lactate deshydrogenase(LDH)in culture media was evaluated.LDH liberation is correlated with the number of lysed cells.Briefly,4000 cells/well were seeded inflat bottom96-well plates(Corning Incor-porated,Corning,NY,USA)and incubated for24h.After HUVEC exposure to the different concentrations of LEH,cells were lysed using a lysis buffer,as described by the manufacturer.LDH activ-ities in the culture media and in the corresponding cell lysates were measured at490nm on a micro-plate reader(Bio-Tek Instru-ments,Winooski,VT,USA).Values were corrected for background absorbance.2.8.1.Assessment of potential residual CH2Cl2cytotoxicityTo ensure that the dichloromethane used in the hemoglobin purification process was not responsible for any cell death,a sample242V.Centis,P.Vermette/Colloids and Surfaces B:Biointerfaces65(2008)239–246 of M-199culture media was treated in the same way.In a con-trol experiment,M-199was exposed to CH2Cl2with subsequentcentrifugation and evaporation procedures(steps2–3,Section2.2).Then,10%(v/v)of de-complemented FBS was added to the media.This“CH2Cl2treated”M-199sample was then added inflat bot-tom96-well plates seeded with4000cells/well.Cells were thenincubated for24h with this M-199and a standard MTS assay wasperformed after the exposure.Normal M-199media was used ascomparison.The absorbance was read at490nm on a micro-platereader(Bio-Tek Instruments,Winooski,VT,USA).Values were onceagain corrected for background absorbance.Results were done intriplicate.2.9.Statistical analysisAll the data collected throughout the study were expressed asmeans±standard deviations(S.D.s).Analysis of variance(ANOVA)was used to determine if data were significantly different usingp≤0.05.3.Results and discussion3.1.Hemoglobin purification3.1.1.SDS-PAGEFig.1shows SDS-PAGE results for the Hb purification proceduredescribed above.In this study,the SDS-PAGE technique was usedas a qualitative method to show the presence of hemoglobin in thesample.Proteins of interest were well defined on the ne Bshows the molecular weight marker containing proteins rangingfrom6.9to210kDa while lanes A and C show purified samples ofhuman Hb and a commercial hemoglobin standard,respectively.It is possible to observe the tetramer,dimer and monomer com-posing hemoglobin at values of,respectively,64,32and16kDa(black arrows on left side,respectively noted1,2and3).The sub-units are visible since the sample was heated prior to its run,thusdenaturing the protein.When purified Hb was compared with thecommercial sample,it was possible to observe the same bandsexcept for one(under the last arrow),which was present only ontheFig.1.SDS-PAGE nes A and C show a purified human hemoglobin sample and a commercial hemoglobin ne B presents broad-range molecular weight markers.Black arrows on left side(1,2,and3)show,respectively,the64,32 and16kDa subunits of the protein.purified sample.It was published by Pearson et al.that this band is not apparent on freshly prepared specimens[28],as opposed to outdated blood,as in our case.Also,the observed streaking in the obtained SDS-PAGE analyses can be caused by the presence of other proteins.Therefore,the effect of the presence of these other proteins on the functionality of Hb has been tested(see below).3.2.Hemoglobin functionalitySpectra of hemoglobin encapsulated in liposomes are presented in Fig.2.Fig.2A clearly shows the difference between oxygen-loaded and unloaded states of the hemoglobin.This difference is even more evident in Fig.2B,showing the characteristic Q-bands and the shift from538and569nm to540and577nm.Panel C of Fig.2summarizes the shifts of hemoglobin from oxygen-loaded states to unloaded ones.Also,Fig.2C shows that hemoglobin can be oxygen-loaded and unloaded several times.The ability of the pro-tein to shift back and forth from one position to another(in thiscase,Fig.2.(A)Complete visible spectra of oxygen-loaded and unloaded purified human hemoglobin,(B)zoom on Q-bands showing the shift between oxygen-loaded and unloaded positions of purified human hemoglobin,and(C)variation of Q-band wavelengths after exposure to carbon monoxide and oxygen.V.Centis,P.Vermette /Colloids and Surfaces B:Biointerfaces 65(2008)239–246243Table 1Mean particle diameter of empty liposomes and liposomes encapsulating hemoglobin stored at 4◦C,22◦C and 37◦C over 2weeks4◦C22◦C37◦CEmpty liposomesLiposomes encapsulating hemoglobin Empty liposomes Liposomes encapsulating hemoglobin Empty liposomes Liposomes encapsulating hemoglobin Day 1220(39.6)226(48.5)240(45.6)269(47.2)229(48.1)239(53.3)Day 7229(41.8)237(47.7)245(46.2)252(45.1)249(44.2)255(42.7)Day 14240(40.7)239(57.6)249(45.9)247(47.8)248(45.6)254(50.1)Data in parentheses indicate the %Polydispersity (%Pd).from oxygen to carbon monoxide)demonstrates the functionality of hemoglobin even after its encapsulation into liposomes.3.3.Liposome characterization3.3.1.Particle size distributionResults of size distribution of liposomes encapsulating hemoglobin are presented in Table 1.It is evident from the data that the size distributions were unimodal and relatively narrow for all samples.%Polydispersity (%Pd)values of all LEH disper-sions were higher than 40%(Table 1).Therefore,LEH dispersions can be considered to as polydispersed.The term polydispersity is derived from the polydispersity index (PdI).PdI is a number cal-culated from a simple two-parameter fit to the correlation data called a cumulants analysis.PdI is known as the relative variance,while %Pd is the coefficient of variation or relative polydisper-sity and can be expressed as (PdI)1/2×100(Malvern Instruments,UK).As a rule of thumb,samples with %Pd ≤20%are considered monodisperse.The size of a particle is an important factor in determining its use as an oxygen carrier.Size is of great importance when one wishes to use these particle-systems in a body to supply increased (or opti-mized)oxygen concentration.Rapid extravasion from the body by the reticulo-endothelial system (RES)and immunogenic effects are only a few of the problems foreign particles encounter in the body [8,19,20,29–34].For tissue engineering purposes,particle size is also an important issue to consider.Liposomes added to a culture medium should be able,for example,to circulate through porous scaffolds often used in tissue engineering.Also,as for LEH,their small size (±200nm)should enable to increase the surface area between the liposomes and the dispersing medium phase allowing a better oxygen transfer.The results in Table 1show that LEH sizes are within the expected range of the 200-nm pores of the extrusion membrane used.These results are comparable and consistent with the findings of Arifin and Palmer [35,36].Using asymmetric field flow fraction-ation coupled to a multi-angle static light scattering photometer,they found that liposomes extruded through membranes having 200-nm pore diameter exhibited a mean diameter that was close to that of the membrane.The dispersion observed was unimodal with distribution widths of 10–20nm.Mean LEH diameter does not vary significantly with storage tem-perature (p ≤0.01).Still,LEH samples should be kept at 4◦C prior to their use in order to limit possible particle size enlargement.Stor-age time of 14days affected to some extent particle size growth in the samples (p ≥0.05).On the other hand,all the preparations exhibited a unimodal distribution,allowing to conclude,that over a short period of time (≤7days),the particles should not aggregate or fuse (p ≤0.05).PEG grafted onto the surface of the vesicles cer-tainly help to stabilize LEH by creating a steric hindrance around liposomes [18].The molar ratio of PEG used in our study is fairly small compared to those of other studies in which PEG was used.But this PEG concentration was sufficient to stabilize the liposomes over 14days,a time corresponding to a culture period required to grow a network of vascular micro-vessels,for instance.The data presented in Table 1reveal that the encapsulation of hemoglobin molecules does not seem to affect the size distribution of the liposomes.Liposomes encapsulating hemoglobin exhibit the same size distribution behaviour as “empty”ones [37].3.3.2.Zeta potentialMeasurements of zeta potential can yield information about the colloidal stability of particle dispersions [36–39].On one hand,in a solution containing no proteins,no amino acids,and little to no multi-valent electrolytes,the larger the magnitude of the zeta potential the more stable the dispersion should be.Limited floccu-lation occurs between |5|and |15|mV [37].For “naked”liposomes (prepared in the same manner as LEH,but hydrated with M-199culture media instead of hemoglobin),it was shown that the zeta potential at the surface of liposomes was approximately −45mV [12,40].When PEG 2000was added to the liposome preparation,the zeta potential increased to reach a plateau around −5mV [12].ThisFig.3.TEM images revealing shape,structure,and sizes of liposomes encapsulating hemoglobin molecules.244V.Centis,P.Vermette/Colloids and Surfaces B:Biointerfaces65(2008)239–246increase of the surface charge was thought in part to be because of the drag caused by the presence of PEG chains on the liposome sur-face,decreasing the mobility of the liposomes,hence affecting the zeta potential[12,41].On the other hand,the presence of surface charge should be counter-balanced with a good steric hindrance in solutions containing proteins and amino acids(e.g.,culture media), as these often charged molecules can adsorb on the surface of the particles and enhance particle aggregation.For the PEGylated liposomes encapsulating Hb studied here,the mean zeta potential on the particle surface was−7.16±0.33mV. This indicates that limited aggregation of the particles should be observed and that stability over time can be,to some extent,con-trolled,as observed in Table1.In fact,over14days,the particle size did not vary for a given temperature and confirmed the stability of the distribution.The values obtained can also be compared with results obtained by Sakai et al.[37].They reported LEH,without PEG and PA,with a surface charge of−21mV.PA was reported to decrease surface charge proportionally to its increase in concentra-tion in the lipid bilayer[37,42].3.3.3.pH and osmolarity of culture media with LEHpH and osmolarity of culture media containing either“empty”liposomes or LEH were monitored over24h.All the solutions had a constant pH of7.4.As for osmolarity measurements,for both“empty”liposomes and M-199controls,the values remained constant at approximately330±3mOsm.The osmolarity of LEH dispersions was319±2mOsm.From these data,it can be concluded that the addition of dispersions of“empty”liposomes and LEH does not affect the osmolarity of the culture medium used in this study.3.4.Transmission electron microscopy(TEM)analysesTEM images of LEH are presented in Fig.3.In the TEM images, the characteristic aspects of large unilamellar vesicles(LUV)can be observed[14].Large unilamellar vesicles are liposomal structures exhibiting a rather empty core that can be used to encapsulate different molecules,such as drugs or proteins[15].LUV’s can be prepared from large“onion-like”structures of multilamellar vesi-cles(MLV)by different methods such as extrusion.The TEM images clearly show that the extrusion method was convenient to produce LEHs.The distinctively recognizable double-layered membrane and the empty core are good indications of a LUV structure[13,14].TEM images revealed that LEH(over62individual liposomes)have a mean diameter of170±50nm.These results are in good agreement with the ones obtained from dynamic light scattering analyses. 3.5.Cytotoxicity of liposomes encapsulating hemoglobin(LEH)Fig.4shows the behaviour of HUVECs in contact with disper-sions made of different concentrations of LEH.Similar cytotoxicity trends were observed when the same experiments were conducted using humanfibroblasts from foreskin(data not shown).Fig.4A and B illustrate the percentage of live cells when HUVECs were exposed to LEH dispersions,while Fig.4C shows the percent-age of cell death after similar exposure.Statistical analyses of the results of these three cytotoxicity assays reveal that exposure time had a significant effect on cell viability(p=0.05).The analyses also show that LEH concentration had an effect on the cell death rate.For example,a significant difference(p≤0.05)was observed between 5and1mg/mL for all tested exposure times.From Fig.4,it can be seen that approximately50%cell loss occurred following24-h cell exposure to the LEH dispersions.The results obtained from the three assays are consistent with one another(p≤0.05).Unfor-tunately,these results cannot be directly correlated with thoseof Fig.4.Cytotoxicity assays of HUVECs exposed to PEGylated liposomes containing hemoglobin for different exposure times.(A)AQueous One assay,(B)CyQuant®assay,and(C)lactate deshydrogenase assay.All data are significantly different with p≤0.05.other studies since very few long-term in vitro tests have been car-ried out.A recent study by Yamaguchi et al.[19]investigated the short and long-term effects of hemoglobin vesicles(HbVs)on the clono-genic and proliferative activity of human hematopoietic progenitor cells derived from umbilical cord blood.They found that,at a concentration of3%(v/v),continuous exposure of HbV signif-icantly decreased the number and size of mature-committed colonies[19].Moreover,HbVs also notably reduced the number of high-proliferative potential colony-forming cells and lead to the suppression of cellular proliferation and differentiation in liquid culture[19].On the other hand,the same study presents the effect of HbV exposure to cord blood for20h or3days.Yamaguchi et。

C开头化学英文期刊对应数据库C开头化学英文期刊对应数据库: Canadian Journal of Chemistry2001- (NRC)Canadian Journal of Microbiology2001- (NRC) Carbohydrate Research1995- (Elsevier)Catalysis Communications1995- (Elsevier)Catalysis Letters1997- (Springer-Kluwer)Catalysis Surveys from Asia2003- (Springer-Kluwer) Catalysis Surveys from Japan1997-2002 (Springer-Kluwer) Catalysis Today1995- (Elsevier)Catalysts & Catalysed Reactions (RSC)CATTECH2000-2003 (Springer)Cell and Tissue Banking2000- (Springer)Cellulose1997- (Springer-Kluwer)Cellulose Chemistry and TechnologyCentral European Journal of Chemistry2003- (CESJ) ChemBioChem2000- (Wiley)Chemical Biology Virtual Journal2002- (RSC)Chemical Communications1996- (RSC)Chemical Communications (London)1965-1968 (RSC)The Chemical Educator1997- (Springer)Chemical & Engineering News1998- (ACS)Chemical Engineering Science1995- (Elsevier) Chemical Engineering & Technology1998- (Wiley) Chemical Hazards in Industry1981- (RSC)Chemical Health and Safety1995- (Elsevier) Chemical Market Report1996-Chemical and Petroleum Engineering2000- (Springer) Chemical & Pharmaceutical Bulletin1999-Chemical Physics1995- (Elsevier)Chemical Physics Letters1995- (Elsevier)The Chemical Record2001- (Wiley)Chemical Research in Chinese UniversitiesChemical Research in Toxicology1988- (ACS) Chemical Reviews1924- (ACS)Chemical Science2004- (RSC)Chemical Society Reviews1972- (RSC)Chemical Technology2005- (RSC)Chemie in unserer Zeit2000- (Wiley)Chemistry - A European Journal1998- (Wiley) Chemistry & Biodiversity2004- (Wiley)Chemistry & Biology1995- (Elsevier)Chemistry in Britain2000-2003 (RSC)Chemistry Education Research and Practice2005- (RSC)Formerly:University Chemistry Education1997-2004 (RSC) Chemistry of Heterocyclic Compounds2000- (Springer-Kluwer) Chemistry & IndustryChemistry International1997-Chemistry Letters1997-Chemistry of Materials1989- (ACS) 【简称Chem. Mater.】Chemistry of Natural Compounds2000- (Springer-Kluwer)Chemistry and Physics of Lipids1995- (Elsevier)Chemistry and Technology of Fuels and Oils2000- (Springer-Kluwer) Chemistry World2004- (RSC)Chemometrics and Intelligent Laboratory Systems1995- (Elsevier) Chemosphere1995- (Elsevier)ChemPhysChem2000- (Wiley)Chemtracts2000-Chinese Chemical Letters1999-Chinese Journal of Chemistry中国化学 2000-2004 (有机所)Chinese Journal of Chemistry中国化学 2005- (Wiley))Chinese Journal of Geochemistry1985-Chinese Journal of Organic Chemistry有机化学 2000-Chinese Journal of Polymer Science2000- (Springer)Chinese Journal of Reactive PolymersChinese Science BulletinChimia1997-Chirality1996- (Wiley)Chromatographia2003- (Springer)Collection of Czechoslovak Chemical Communications1994- Colloid Journal2000- (Springer-Kluwer)Colloid & Polymer Science1998- (Springer)Colloids and Surfaces A: Physicochemical and Engineering Aspects1995- (Elsevier)Colloids and Surfaces B: Biointerfaces1995- (Elsevier)Combinatorial Chemistry - an Online Journal2003- (Elsevier) Comptes Rendus de l'Academie Bulgare des SciencesComptes Rendus de l'Académie des Sciences - Series IIB - Mechanics-Physics-Chemistry-Astronomy1995- (Elsevier) Comptes Rendus de l'Académie des Sciences - Series IIC - Chemistry1998-2001 (Elsevier)Comptes Rendus Chimie2002- (Elsevier)Computational and Theoretical Polymer Science1997-2001 (Elsevier)Computational Biology and Chemistry2003- (Elsevier)Computers & Chemistry1995-2002 (Elsevier)Computing and Visualization in Science1997- (Springer)Concepts in Magnetic Resonance Part A1997- (Wiley)Concepts in Magnetic Resonance Part B: MagneticResonance Engineering2001- (Wiley)Contemporary Organic Synthesis1994-1997 (RSC)Continuum Mechanics and Thermodynamics1995- (Springer) Contributions to Mineralogy and Petrology1995- (Springer) Coordination Chemistry Reviews1995- (Elsevier)Critical Reviews in Analytical Chemistry2003- (Elsevier)Croatica Chemica Acta1996-Crystal Growth & Design2001- (ACS)Crystal Engineering1999- (Elsevier)CrystEngComm1999- (RSC)Current Biology1995- (Elsevier)Current Medicinal Chemistry2000- (Bentham) (下载原文与图书馆联系)Current Medicinal Chemistry - Anti-Cancer Agents2001- (Bentham) (下载原文与图书馆联系)Current Opinion in Biotechnology1995- (Elsevier)Current Opinion in Cell Biology1995- (Elsevier)Current Opinion in Chemical Biology1997- (Elsevier)Current Opinion in Colloid & Interface Science1999- (Elsevier) Current Opinion in Genetics & Development1995- (Elsevier) Current Opinion in Pharmacology1995- (Elsevier)Current Opinion in Plant Biology1995- (Elsevier)Current Opinion in Structural Biology1995- (Elsevier)Current Organic Chemistry2000- (Bentham) (下载原文与图书馆联系)Current T opics in Medicinal Chemistry2001- (Bentham) (下载原文与图书馆联系)。

Available online at Colloids and Surfaces A:Physicochem.Eng.Aspects317 (2008) 551–556An environmental scanning electron microscopy examination of thefilm formation mechanism of novel acrylic latexKalin I.Dragnevski∗,Athene M.DonaldSector of Biological&Soft Systems,Department of Physics,Cavendish Laboratory,University of Cambridge,J J Thomson Avenue,Cambridge CB30HE,UK Received27July2007;received in revised form20November2007;accepted23November2007Available online 4 December 2007AbstractWe have employed environmental scanning electron microscopy(ESEM)to study thefilm formation mechanisms of two acrylic latex compo-sitions,here defined as standard(carboxymethyl cellulose stabilised)and novel(stabilised with a novel polysaccharide derived from agricultural waste).The ESEM analysis revealed that the microstructure of the standard system consists of individual particles and upon evaporation a contin-uousfilm is formed,which is consistent with the current models.However,in the case of the novel system the microstructure consists of individual particles and clusters and during evaporation a discontinuousfilm is formed with voids present within its structure.Based on the experimental evidence we propose a modification to thefilm formation mechanism for the novel latex system.© 2007 Elsevier B.V. All rights reserved.Keywords:Polymer latex;Film formation;ESEM1.IntroductionPolymer lattices,with their wide range of applications,have been the subject of many theoretical and experimental stud-ies.When used for its traditional applications,i.e.as paint or adhesive,the latex is applied in its wet state to a surface and allowed to dry andfilm form under ambient conditions.There-fore,conventional electron microscopy,with its extreme drying and sample preparation requirements,will not be suitable for the examination of lattices in their natural wet state.On the other hand,environmental scanning electron microscopy(ESEM)[1], which offers the possibility to image‘wet’and insulating spec-imens,has been successfully used in the study of a number of systems and dynamic processes including lattices andfilm formation[2–7].ESEM is based on the use of a multiple aperture graduated vacuum system,which allows specimens to be imaged under water vapour or other auxiliary gases,such as nitrogen or nitrous oxide[4].In this way,the chamber can be held at pressures usu-ally in the range of1–10Torr[8],while the gun and column remain at pressures of∼7.5×10−7Torr.Moreover,by using a ∗Corresponding author.E-mail address:kd281@(K.I.Dragnevski).correct pumpdown procedure[9]and by controlling the temper-ature of the specimen,which in the ESEM is usually done by using a Peltier stage,dehydration can be inhibited and hence samples can be imaged in their‘natural state’.Furthermore,by taking into consideration the saturated vapour pressure(SVP) curve for water as a function of temperature[9]and by increas-ing the temperature of the specimen or reducing the chamber pressure,it is possible to produce evaporation conditions within the specimen chamber,which allows examination of the process offilm formation.As mentioned above,polymer lattices are important indus-trial products and the subject of many research tex, which is an example of a wet insulating material,can be defined as a colloidal suspension of spherical polymer particles with varying diameters.When water is allowed to evaporate from the system,the aqueous suspension undergoes a series of trans-formations,which result in the formation of a continuous dry polymerfilm.This process,known asfilm formation,contains four main stages that can be described as follows[10–18]—stage I:dispersed suspension of polymer particles;stage II:con-centrated suspension of particles in contact with each other, surrounded by solvent-filled interstices;stage III:ordered array of deformed particles;stage IV:a molecularly continuous and homogeneousfilm formed as a result of polymer interdiffu-sion.0927-7757/$–see front matter© 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.colsurfa.2007.11.042552K.I.Dragnevski,A.M.Donald/Colloids and Surfaces A:Physicochem.Eng.Aspects317 (2008) 551–556Fig.1.Schematic representation of an idealizedfilm formation process.Adapted from Keddie et al.to include the intermediate stage II*.Keddie et al.[5]used environmental scanning elec-tron microscopy and multiple-angle-of-incidence ellipsometry (MAIE)in the study of latexfilm formation.They concluded that an intermediate stage,between II and III,has been omitted in the conventional descriptions[10–18].The stage,defined as II*,is characterized by a randomly packed array of deformed particles which still contain water-filled interstices.A schematic representation of the process is shown in Fig.1.More recently,Keddie and co-workers[19,20]investigated the possibility of creating heterogeneousfilms,by mixing car-bon nanotubes(CNTs)with waterborne polymer particles.It was found that the mechanical properties of the nanocomposite coatings can be greatly improved,while maintaining their opti-cal clarity.However,it is important to note that all of the above studies were carried out using continuous polymerfilms.In recent years,environmental considerations have provided another strong motivation for developing coatings from renew-able resources and eliminating coalescing aids[21].These volatile organic components(VOCs)temporarily lower the glass transition temperature(T g)of the latex,which provides mobility to the polymer chains,thus allowing easier particle deformation and interdiffusion.Possible ways of eliminating VOCs,and pro-ducing lattices with lower T g,include blending hard and soft particles[22–24]so that the soft particles deform around the harder ones.However,in this case it is important to avoid seg-regation within the dispersion,ensuring an even distribution of particles throughout thefilm.Alternatively,core–shell particles, with a soft deforming shell surrounding a hard core,can be used for avoiding the issues with phase separation[25,26].In this paper we present the results from an ESEM investi-gation into thefilm formation mechanisms of a novel acrylic latex that has been stabilised by using a new polysaccharide, derived from agricultural waste and a standard polymer sys-tem,where the conventional carboxymethyl cellulose(CMC) has been used as a stabiliser.The novel polysaccharide con-sists of a number of monosaccharides(including arabinose and xylose)formed fromfive-and six-membered rings and has a low molecular weight,only a few thousand a.m.u.s rather than the hundreds of thousands found in cellulose for example.The polysaccharide also contains a significant amount of interfacially active protein∼15%.It is suggested that the initial latex particle stabilization comes from the protein component and ultimately the polysaccharide component stabilises the latex particles by adsorbing on their surface,rather than by chemically grafting on the growing polymer particles,which is the case for the con-ventionally used CMC.Initial examinations[27]have indicated that the novel latex canfilm form without the addition of coa-lescing solvents,which as suggested above,on one hand would provide an alternative method for the production of VOC-free architectural coatings and on the other would comply with the stringent EU and DEFRA regulations[28].2.Materials and methodsTwo aqueous latex compositions,supplied by ICI Plc,based on copolymers of methyl methacrylate(MMA)and2-ethylhexyl acrylate(2-EHA)were studied.In this paper the latex stabilised with the conventionally used by ICI Plc carboxymethyl cellulose will be referred to as‘standard’and the other stabilised with the new polysaccharide as‘novel’.The two lattices were initially about55wt.%polymer.The glass transition temperatures of the two lattices were determined by differential scanning calorime-try(DSC),carried out on dry specimens,using a PerkinElmer Pyris1instrument.The measured temperatures were279.8K for the standard and280.1K for the novel latex.The minimum film formation temperatures(MFFT)of the two lattices were measured by using a MFFT-Bar and were found to be278K and 279K,respectively.The microstructural analysis was carried out on an FEI XL-30environmental scanning electron microscope equipped with a Peltier stage.Wet samples from the above formulations were placed onto the cooling stage in the microscope chamber at a temperature of ca.274K.An evaporation-inhibiting pumpdown sequence was then performed,with the ambient air progres-sively replaced by water vapour.Once the purging cycle was completed,water vapour pressures and working distances of 3.5–4.5Torr and9.5–11.5mm were set,which provided suitable imaging environments.Imaging of the specimens was carried out at an accelerating voltage of10kV.Previous studies[2–5,29] have shown that the use of moderate beam voltages in combina-tion with fairly high pressures results in minimal beam damage, which proved to be the case in this study.Increasing the temper-ature of the specimens by1◦or2◦above the starting temperature of274K,as explained above,resulted in further dehydration of the lattices,which allowed examination of the process offilm formation.3.Results and discussionThe results of the microstructural observations are presented here.Firstly,thefilm formation mechanism of the standard latex will be considered.This will be followed by a presentation of the results for the novel polymer system.However,prior to considering the above,it is important to note that when we refer to lattices as being‘wet’,some water has in actual fact been removed from the surface of the specimens in order to obtain better quality images.Keddie et al.[2,5]used a simi-lar approach in the study of latexfilm formation by means of ESEM.It was found that despite the fact that some of the sur-K.I.Dragnevski,A.M.Donald /Colloids and Surfaces A:Physicochem.Eng.Aspects 317 (2008) 551–556553Fig.2.ESEM micrograph showing the surface of a standard latex specimen in stage II/II*.Imaging conditions:T =274K;p =3.9Torr.face water had been removed,the bulk of the samples remained ‘wet’.3.1.Standard latexFrom the ESEM image of the standard latex (Fig.2),it can be seen that under ‘wet’conditions the microstructure of the speci-men consists mainly of randomly distributed individual particles with an average size of ca.300nm.This was also confirmed by particle size measurements (Fig.3)carried out on a Coulter LS230Light Scattering Apparatus.Due to the fact that some of the water has already been removed,as explained above,some of the polymer particles are in contact.Despite that,they are still physically distinct,i.e.no significant deformation has occurred,and therefore it can be concluded that the latex is in stage II/II*.Fig.4reveals the surface microstructure of a standard latex specimen at a temperature of 276K.It is clearly seen that,at this slightly increased temperature,water evaporation has taken place,which results in the formation of a continuous polymer film.However,due to the fact that not all particles have lost their identity and some boundaries are still clearly visible,itcanFig.3.Particle size distribution of the standard latex,confirming the fact that the microstructure of the specimens in the ‘wet’state consists predominantly of individual polymer particles with sizes in the range of 300nm.Fig.4.ESEM micrograph of a standard latex specimen in stage III/IV .Imaging conditions:T =276K;p =3.9Torr.be concluded that under these conditions the latex is in stage III/IV .Due to the fact that imaging of the latex specimens was carried out below their T g of 279.8K,it is somewhat surpris-ing to observe the latter stages of film formation.However,it is suggested that as the microstructural analysis was carried out at temperatures very close to the minimum film formation tem-perature,partial particle deformation and coalescence,would naturally be expected to take place.It was also found that further increases in temperature did not lead to the observation of other surface morphologies.Based on the above results,which are comparable to those obtained in previous studies [1–5],it can be said that the film formation mechanism of the standard acrylic latex is in a good agreement with the conventional descriptions.3.2.Novel latexFig.5depicts the surface morphology of a novel latex spec-imen under ‘wet’conditions.The microstructure appears to be similar to one observed for the standard latex.The individual polymer particles,although in contact,are physicallydistinctFig.5.ESEM micrograph of a novel latex specimen in stage II/II*.Imaging conditions:T =274K;p =3.9Torr.554K.I.Dragnevski,A.M.Donald /Colloids and Surfaces A:Physicochem.Eng.Aspects 317 (2008) 551–556and therefore it can be concluded that the latex is in stage II/II*of the film formation process.However,another interesting feature that can be seen at slightly higher magnifications within the microstructure of the novel latex specimens (Fig.5inset )is the presence of a large number of clusters with sizes in the range 2–5m.The presence of these clusters was also confirmed by AFM [27]and particle size measurements (Fig.6a and b)carried out by ICI Plc using a Coulter LS230Light Scattering Apparatus.From the above ESEM images it is also evident that the clus-ters seen within the structure of the novel latex appear to have been formed by aggregation,followed by partial coalescence of individual particles,rather than complete coalescence.Here,partial coalescence is defined as the process of formation of agglomerates of spherical particles that are physically distinct,whereas complete coalescence is termed to be the formation of a featureless polymer structure.The factors leading to the formation of these clusters are currently being investigatedandFig.6.Particle size distribution (a)and AFM image (b)of the novel latex system,confirming the presence of a large number of clusters with sizes in the range of 2–5m.are believed to be related to the low molecular weight of the polysaccharide and its branched molecular structure [27].Further dehydration of the specimens resulted in the for-mation of a discontinuous film,with voids present within its structure (Fig.7).Similar to the standard latex,under these conditions not all particles and/or clusters appear to have com-pletely lost their identity and therefore it can be concluded that the latex is in stage III/IV of the film formation process.At this point,it is important to note that just as with the standard latex,the examination was carried out at temperatures close enough to the minimum film formation temperature of the latex to allow observation of the latter stages of the film formation process.The discontinuity of the film can be explained by taking into consideration the shape and size of the clusters.As seen from the ESEM results,the aggregates of spherical particles have differ-ent sizes and shapes.Therefore,it can be expected that during water evaporation,i.e.when clusters and individual particles come in contact,voids within the polymer film would easily form.It is believed that the presence of the clusters,which are formed during the latex synthesis,reduces the need for coalesc-ing solvent as part of the film formation has been achieved before film lay down and evaporation of the water.Thus,the creation of this partially coalesced polymer network during evaporation results in the formation of a film,which is in the middle of the two extremes,i.e.full coalescence and no coalescence.In summary,it can be said that despite the fact that the mechanism of cluster formation in the novel acrylic latex is yet to be fully revealed,the film formation process that the system undergoes as a result is clearly seen to be different from the standard one.Although,the overall process appears to be similar,some modifications of the individual stages of the conventional model need to be made,in the case of the novel latex,due to the presence of clusters in the starting material.The stages that the system undergoes during water evaporation can be described as follows—stage I:dispersed suspension of polymer particles and clusters,formed by aggre-gation of individual particles;stage II:concentratedsuspensionFig.7.ESEM image of a novel latex specimen in stage III/IV .Imaging condi-tions:T =276K;p =3.9Torr.K.I.Dragnevski,A.M.Donald/Colloids and Surfaces A:Physicochem.Eng.Aspects 317 (2008) 551–556555Fig.8.Schematic representation of thefilm formation mechanism for the novel latex system.of particles and clusters in contact with each other,surrounded by solvent-filled interstices;stage II*:randomly packed array of deformed particles and clusters that still contain water-filled interstices;stage III:ordered array of deformed particles and clusters with voids present in the structure;stage VI:a dis continuousfilm formed as a result of polymer interdiffusion.A schematic diagram of the process described above is shown in Fig.8.4.ConclusionsEnvironmental scanning electron microscopy has proven to be a successful method for studying the process of evolution of an aqueous polymer dispersion into a polymerfilm.The ESEM results,which are consistent with AFM data obtained by ICI[27],revealed that there are differences in both the microstructures and the drying behaviour of the studied latex systems.In the case of the standard latex,the microstruc-ture mainly consists of individual particles,whereas in the case of the novel latex,the microstructure appears to consist of individual particles and clusters with sizes in the range of 2–5m.Furthermore,during water evaporation,in the case of the standard system a continuousfilm is formed,which is con-sistent with the classical descriptions,whereas in the novel one thefilm formed,is discontinuous,with voids present within its structure.Based on the experimental results obtained in this study we have proposed a modification to the currently accepted mechanism for the formation of polymerfilms during drying.AcknowledgementsThe authors would like to acknowledge ICI Plc for funding this work and providing the latex specimens.We also thank Drs Simon Davies,Phil Taylor,Liz Bone,Martin Murray and Mervin Shannon for useful discussions and advice.References[1]G.Danilatos,Review&outline of environmental SEM at present,J.Microsc.162(1991)391–402.[2]J.L.Keddie,P.Meredith,R.A.L.Jones, A.M.Donald,Film for-mation of acrylic lattices with varying concentrations of non-film forming latex particles,Langmuir12(16)(1996)3793–3801.[3]C.He,A.M.Donald,Morphology of core–shell polymer lattices duringdrying,Langmuir12(26)(1996)6250–6256.[4]P.Meredith,A.M.Donald,Study of wet polymer systems in an environ-mental SEM:some imaging considerations,J.Microsc.181(1)(1996) 23–35.[5]J.L.Keddie,P.Meredith,R.A.L.Jones,A.M.Donald,Kinetics offilmformation in acrylic lattices studied with multiple-angle-of-incidence ellipsometry and environmental SEM,Macromolecules28(1995)2673–2682.[6]A.M.Donald,C.He,P.Royall,M.Sferrazza,N.A.Stelmashenko,B.L.Thiel,Applications of environmental SEM to colloidal aggregation and film formation,Colloids Surf.A:Physicochem.Eng.Aspects174(2000) 37–53.[7]A.Bogner,G.Thollet, D.Basset,P.-H.Jouneau, C.Gauthier,WetSTEM:a new development in environmental SEM for imaging nano-objects included in a liquid phase,Ultramicroscopy104(2005)290–301.[8]D.J.Stokes,Recent advances in electron imaging,image interpretation andapplications:environmental scanning electron microscopy,Phil.Trans.R.Soc.Lond.,A361(2003)2771–2787.[9]R.E.Cameron, A.M.Donald,Minimising sample evaporation in theenvironmental scanning electron microscope,J.Microsc.173(1994) 227.[10]G.L.Brown,Formation offilms from polymer dispersions,J.Polym.Sci.22(1956)423.[11]J.W.Vanderhoff,Mechanism offilm formation of lattices,Chem.ProcessEng.51(5)(1970)89.[12]S.S.V oyutskii,tinova,Role of autohesion duringfilm formationof latex,J.Adhes.9(1977)39.[13]D.P.Sheettz,Formation offilms by drying of latex,J.Appl.Polym.Sci.9(1965)3759–3773.[14]E.M.Boczar,B.C.Dionne,Z.Fu,A.B.Kirk,P.M.Lesko,A.D.Koller,Spectroscopic studies of polymer interdiffusion duringfilm formation, Macromolecules26(1993)5772.[15]M.A.Winnik,Latexfilm formation,Curr.Opin.Colloid Interf.Sci.2(2)(1997)192–199.[16]J.L.Keddie,Film formation of latex,Mater.Sci.Eng.R21(3)(1997).[17]A.F.Routh,W.B.Russel,A process model for latexfilm formation:limit-ing regimes for individual driving forces,Langmuir15(22)(1999)7762–7773.[18]P.A.Steward,J.Hearn,M.C.Wilkinson,An overview of polymer latexfilm formation and properties,Adv.Colloid Interf.Sci.86(3)(2000)195–267.[19]P.Vandervorst, C.H.Lei,Y.Lin,O.Dupont, A.B.Dalton,Y.P.Sun,J.L.Keddie,Thefine dispersion of functionalised carbon nan-otubes in acrylic latex coatings,.Coat.75(2)(2006)91–97.[20]T.Wang, C.H.Lei, A.B.Dalton, C.Creton,Y.Lin,K.A.S.Fer-nando,M.Manea,J.M.Asua,J.L.Keddie,Waterborne nanocomposite pressure sensitive adhesives with high tack energy,optical trans-parency and electrical conductivity,Adv.Mater.18(20)(2006) 2730.[21]M.A.Winnik,J.Feng,Latex blends:an approach to zero VOC coatings,J.Coat.Technol.68(852)(1996)39–50.[22]S.T.Eckersley,B.J.Heimer,Mechanistic considerations of particle sizeeffects onfilm properties of hard/soft latex blends,J.Coat.Technol.69 (864)(1997)97–107.[23]A.Tzitzinou,J.L.Keddie,J.M.Geurts,A.C.I.A.Peters,R.Satguru,Filmformation of latex blends with bimodal particle size distributions:con-556K.I.Dragnevski,A.M.Donald/Colloids and Surfaces A:Physicochem.Eng.Aspects 317 (2008) 551–556sideration of particle deformability and continuity of the dispersed phase, Macromolecules33(7)(2000)2695–2708.[24]S.Lepizzera,C.Lhommeau,G.Dilger,T.Pith,mbla,Film formingability and mechanical properties of coalesced latex blends,J.Polym.Sci., Part B:Polym.Phys.35(1997)2093–2101.[25]D.Juhue,ng,Film formation from dispersion of core shell latexparticles,Macromolecules28(1995)1306–1308.[26]L.Dong,Y.Tong,Y.An,H.Tang,Y.Zhuang,Z.Feng,Study of the blendscontaining core–shell latex polymer,Eur.Polym.J.33(4)(1997)501–503.[27]ICI Plc,Private communication,2006.[28]EU Directive2004/42/CE.[29]Royall,P.,The behaviour of silica in matt water-based lacquers,PhD thesis,University of Cambridge,2000.。

Colloids and Surfaces A:Physicochem.Eng.Aspects 471(2015)45–53Contents lists available at ScienceDirectColloids and Surfaces A:Physicochemical andEngineeringAspectsj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /c o l s u r faWater-soluble complexes of hydrophobically modified polymer and surface active imidazolium-based ionic liquids for enhancing oil recoveryShaohua Gou a ,b ,∗,Ting Yin b ,Liwei Yan b ,Qipeng Guo c ,∗∗aState Key Laboratory of Oil and Gas Reservoir Geology and Exploitation,Southwest Petroleum University,Chengdu 610500,PR ChinabOil &Gas Field Applied Chemistry Key Laboratory of Sichuan Province,School of Chemistry and Chemical Engineering,Southwest Petroleum University,Chengdu 610500,PR China cPolymers Research Group,Institute for Frontier Materials,Deakin University,Locked Bag 2000,Geelong,Victoria 3220,Australiah i g h l i g h t s•A series of copolymer/ionic liquidscomplexes (PAAD/ILs)were used for EOR.•PAAD/C 8mimBr complex can effec-tively reduce the IFT of water/crude oil.•PAAD/C 8mimBr complex with NaCl can further reduce IFT of water/crude oil.•PAAD/C 8mimBr complex exhibits excellent temperature resistance.•PAAD/C 8mimBr complex can enhance oil recovery as high as 21.65%.g r a p h i c a la b s t r a cta r t i c l ei n f oArticle history:Received 20October 2014Received in revised form 27January 2015Accepted 2February 2015Available online 16February 2015Keywords:Ionic liquidsHydrophobically associating copolymer Interfacial tensionEnhancing oil recoverya b s t r a c tThe current study introduces the water-soluble complexes containing hydrophobically associating copolymer and a series of surface activity imidazolium-based ionic liquids (C n mimBr,n =6,8,10,12,14and 16).The polymer,denoted as PAAD,was prepared with acrylamide (AM),acrylic acid (AA)and N ,N -diallyl-2-dodecylbenzenesulfonamide (DBDAP).And the hydrophobic associative behavior of PAAD was studied by a combination of the pyrene fluorescence probe and viscosimetry.Incorporation of C n mimBr (n =10,12,14and 16)in PAAD leaded to the white thick gel,while the pellucid solutions were obtained in complexes of PAAD and C n mimBr (n =6and 8);addition of C 6mimBr around critical micelle concen-tration resulted in a large decrease in viscosity of solution.Therefore,we particularly investigated the performance of PAAD/C 8mimBr complex.The interfacial tension of PAAD/C 8mimBr complex solution and crude oil under different conditions was examined.Moreover,PAAD/C 8mimBr complex exhibited∗Corresponding author at:School of Chemistry and Chemical Engineering,South West Petroleum University,Xindu Avenue 8#,Xindu,Chengdu 610500,Sichuan,PR China.Tel.:+8602883037301;fax:+8602883037333.∗∗Corresponding author at:Polymers Research Group,Institute for Frontier Materials,Deakin University,Locked Bag 2000,Geelong,Victoria 3220,Australia.E-mail addresses:shaohuagou@ (S.Gou),qguo@.au (Q.Guo)./10.1016/j.colsurfa.2015.02.0220927-7757/©2015Elsevier B.V.All rights reserved.46S.Gou et al./Colloids and Surfaces A:Physicochem.Eng.Aspects471(2015)45–53superior temperature resistance and shear reversible performance for enhancing oil recovery(EOR)byrheological test.The promising EOR of21.65%can be obtained by PAAD/C8mimBr complex showing highpotential to utilize this kind of new complex in EOR processes.©2015Elsevier B.V.All rights reserved.1.IntroductionIn fact,with the recovery of the reservoirs all over the world, most crude oil is trapped in the reservoirs after using the conven-tional oil production methods.High world energy demands make the efficient enhancing oil recovery(EOR)techniques have never become as urgent as today[1].Generally,chemical enhancing oil recovery methods are of specific concern in oil recovery,like poly-merflooding,surfactantflooding and polymer–surfactantflooding [2–4].One of the most promising chemical EOR techniques is the polymer–surfactantflooding.The main mechanism of this method is based on the large mobility ratio and the low interfacial tension between the displacementfluid and crude oil.Generally,a lager capillary number(Nc)and/or a lower mobility ratio(M)result in a higher oil recovery,and the most effective way of increasing the Nc is reducing interfacial tension between the displacementfluid and crude oil[1].Hydrophobically associating polymer is a special kind of water-soluble polymer which contains a small amount of hydrophobic monomer[5].This kind of polymer has received increasing atten-tions on account of its unique rheological performance[6].Due to the hydrophobic groups it can generate the intramolecular and intermolecular hydrophobic microarea leading to a considerable increase of viscosity,consequently improving the mobility ratio (M).It has been demonstrated that the performance can be notably changed by combinations of this polymer solution with a certain amount of surfactant[7–9].Although the hydrophobically asso-ciating polymer–surfactantflooding technique is promising,its application to date has been limited due to the rheology perfor-mance of system and the failure in function of surfactant under the reservoir conditions such as poor salt tolerance of anionic surfactant[10].For these reasons,there is growing interest infind-ing a new hydrophobically associating polymer–surfactant system whose properties bestfit the EOR requirements.Ionic liquids(ILs)are liquids at ambient that have unique fea-ture such as high thermal stability,negligible vapor pressure,and favorable chemical stability[11,12].Recently,the incorporation of ionic liquid into polymers has attracted much interest,such as poly-mer/ionic liquid gel membranes with high ionic conductivity and mechanical stability[13],and the thermodynamic phase behav-ior of polymer solution in the presence of different kinds of ILs [14,15].The surface active ILs,imparts them unique physicochemical properties:analogous to common surfactants,they have surface activity.It seems to be a few investigations on the interfacial ten-sion(IFT)of ILs solution/oil system were reported[10,16–21].Those authors recognized its desired behavior at high salinity and tem-perature.Imidazolium based surface active ILs are readily available in technical quantities and these kinds of ILs are one of the com-mon ILs among the ILs families[22,23].Hezave et al.[18]examined the IFT of1-dodecyl-3-methylimidazolium chloride with crude oil under different conditions and performed the coreflooding experiments.They found promising results of both enhanced oil recovery efficiency and adsorption on the rock surfaces.Above investigations show high potential to utilize imidazolium based ILs to replace the traditional surfactants in EOR processes to reduce the IFT.However,few studies are available on incorporation of these surface active ILs into hydrophobically associating polymer for EOR.Based on thesefindings,we report a study on the complexes of the long-chain imidazolium based surface active ILs C n mimBr (n=6,8,10,12,14,and16)and hydrophobically modified polymer denoted as PAAD which was prepared by acrylamide(AM),acrylic acid(AA)and N,N-diallyl-2-dodecylbenzenesulfonamide(DBDAP). According to our previous work[24],the introduction of sulfo-namide structure and aromatic ring can improve the rigidity of the polymer chains exhibiting high-temperature resistance,and based on this,DBDAP containing above structures and long chain structure was designed to prepare the hydrophobically associating polymer to further improve the performance of the polymer.ILs can interact with this polymer by electrostatic force and they can also form micelle-like clusters associated with the polymer hydropho-bic plementary hydrophobic associative behavior data of PAAD obtained by pyrene probefluorescence and viscosimetry were also presented.The IFT of PAAD/C8mimBr complex solution and crude oil under different conditions was measured,and the rheological behavior of the complex was also investigated.TG-DSC was also carried out to study the thermal decomposition of the complex.Moreover,the coreflooding test was conducted.2.Experimental2.1.MaterialsAcrylamide(AM),acrylic acid(AA),dodecylbenzene sulfonic acid(DB),thionyl chloride(SOCl2),diallylamine(DAP),nonaphenol polyethyleneoxy(10)ether(OP-10),N-methylimidazole(mim),1-bromobutane,1-bromohexane,1-bromooctane,1-bromodecane, 1-bromododecane,1-bromotetradecane,1-bromohexadecane, cetyltriethylammonium bromide(CTAB),triethylamine(Et3N), dichloromethane(CH2Cl2),trichloromethane(CHCl3),ethyl acetate,diethyl ether,ammonium persulfate((NH4)2S2O8), sodium bisulfite(NaHSO3),NaCl,and NaOH etc.are all provided by Chengdu Kelong Chemical Reagent Factory,Sichuan.These chemicals are chemically pure or above.CHCl3,ethyl acetate, Et3N and diethyl ether were dried using anhydrous sodium sulfate before used,and other chemicals were used as commercial without further purification.2.2.Synthesis of DBDAPN,N-Diallyl-2-dodecylbenzenesulfonamide(DBDAP)was pre-pared referring to the traditional methods[25].Briefly,dode-cylbenzenesulfonyl chloride was prepared by DB with excess SOCl2under reflux at50◦C for5h.And the reaction of dode-cylbenzenesulfonyl chloride and DAP using Et3N as acid binding agent in CH2Cl2was performed at0–5◦C for6h.The product was washed three times with1wt%diluted hydrochloric acid, 1wt%sodium hydroxide and saturated salt water,respectively, and then the solvent was removed under a vacuum.Obtained DBDAP was brown liquid with a yield of92%.DBDAP:1H NMR (400MHz,CDCl3):ı=7.76(d,2H,J=7.2Hz,Ar H),7.07(d,2H, J=8.0Hz,Ar H),5.77–5.81(m,2H,CH2C H CH2), 5.13–5.32 (m,4H,C H2CH CH2),3.15–3.19(m,4H,SO2N(C H2)2),2.46 (t,2H,J=8.0Hz,Ar C H2),1.47–1.57(m,2H,Ar CH2C H2), 1.20–1.24(m,18H,Ar CH2CH2(C H2)9CH3),and0.87(t,3H, J=4.0Hz,C H3CH2),ppm.S.Gou et al./Colloids and Surfaces A:Physicochem.Eng.Aspects471(2015)45–5347Scheme1.The synthetic process of PAAD.2.3.Synthesis of PAADPreparation of PAAD was conducted via free radical copolymer-ization of AM,AA and DBDAP in aqueous solution with emulsifier OP-10.DBDAP(0.02g),AM(6g),AA(4g)and OP-10(0.1g)were dissolved in40mL deionized water with a magnetic stir bar,and the pH was adjusted around7using1.0mol/L NaOH solution. Then,(NH4)2S2O8(0.0368g)and NaHSO3(0.0132g)were added in at40◦C for8h under N2atmosphere.The resulting product was obtained by repeatedly washed with ethanol and dried at40◦C,and then kept in a desiccator.The synthetic process of PAAD is shown in Scheme1.2.4.Synthesis of PAAD/ILs complexSurface active ILs,C n mimBr,n=6,8,10,12,14and16,were prepared and purified as reported in literature[23,26].The water content of ILs is controlled by drying them at100◦C under vacuum conditions.A desired amount of PAAD was dissolved in distilled water under mechanical stirring until a clear homogeneous solu-tion was obtained.Then,the above ILs with definite concentration were added to the prepared polymer solution at50◦C for4h. Finally,the complex solutions of polymer and different ILs were obtained.2.5.CharacterizationFTIR spectra were determined with the KBr pellets method using WQF-520Fourier transform infrared spectrometer in the optical range400–4000cm−1by the averaging of32scans(Bei-jing Rayleigh Analytical Instrument Corporation,China).1H NMR spectra were recorded on a Bruker AV III-400NMR spectrometer (Bruker,Switzerland)in D2O or CDCl3.The intrinsic viscosity of copolymer was measured with a Ubbe-lohde viscometer using1mol/L NaCl aqueous solution as the solvent with the dilution extrapolation method at30.0±0.1◦C, and the initial concentration of copolymer was0.001g/mL (C0=0.001g/mL).The viscosity-average molecular weight of copolymer can be calculated from the intrinsic viscosity value by employing Mark–Houwink equation.However,it should be pointed out that this measurement is an approximate and relative method on the determination of the viscosity-average molecular weight of hydrophobically associating polymers due to the effect of intra-molecular hydrophobic interaction.2.6.Apparent viscosity measurementThe apparent viscosity of different solutions was obtained on a Brookfield D-III+Pro viscometer(Brookfield,USA)with different viscometer rotors0#(6.0rpm)62#(18.8rpm)and63#(27.3rpm).2.7.Pyrenefluorescence probeThefluorescence intensities of copolymer were measured with a Shimadzu RF-5301PC Fluorescence spectrophotometer with excitation at335nm,with a slit width of5nm and in a spectral range350–550nm.The different concentrations of copolymer solu-tions with pyrene were prepared with redistilled water,and the concentration of pyrene was about1.25×10−6mol/L.The ratios (I1/I3)of the strength of thefirst peak to that of the third peak in fluorescence spectra were calculated.2.8.Thermogravimetry and differential scanning calorimetryThe water of PAAD/C8mimBr complex solution with a cer-tain mass ratio was removed through rotary evaporation to test with thermogravimetry and differential scanning calorimetry(TG-DSC)using a STA449F3synchronous thermal analyser(Netzsch, Germany)in the temperature range40–700◦C at a heating rate of 10◦C/min under air atmosphere.2.9.Rheological experimentsThe effect of temperature on the viscosity of samples was mea-sured by HAAKE RS600Rotational Rheometer(HAAKE,Germany) at shear rate of170s−1to simulate injection rate at the heating rate of3◦C/min from30to120◦C.The shear thinning behavior of samples was performed in the range of2–500s−1shear rates at 30◦C.2.10.Interfacial tension testSurface tension measurements were performed with TX500C SpinningDrop Interface tensiometer(CNG USA Co.)using the drop volume method at30◦C.The oil used in interfacial tension test is prepared by crude oil and kerosene with a mass ratio of2:1,and the density is0.8982g/cm3.The crude oil sample is obtained from Bohai Suizhong Oilfield(SZ36-1CEPK).Then,the interfacial tension was measured applying a rotating velocity of5000rpm.The density of each system was measured.2.11.Coreflooding testCoreflooding test was using stainless steel packed with sand (30cm in length and2.55cm in diameter,approximately),and the size distribution of sand was80–100items.The apparent viscos-ity of simulated crude oil was30.6mPa s at70◦C.NaCl solution was injected in core until a steady pressure to obtain the porosities of core by gravimetry,and permeability was obtained by injecting NaCl solution at a constant rate of9.99mL/min using Darcy’s law [27].The sand with crude oil has been saturated at0.1mL/min at 70◦C for96h,and oil saturation was calculated[4].Firstly,the waterflooding was conducted with the NaCl solution until water cut reached at95%,and then it wasflooded with0.2PV cumulative injection volume of chemicals.Finally,the extrapolated waterflooding was conducted with the NaCl solution to obtain water cut95%once more.The injection rate was0.3mL/min in flooding process.The oil recovery was determined as the following equation:EOR=E−E W(1)48S.Gou et al./Colloids and Surfaces A:Physicochem.Eng.Aspects 471(2015)45–53Table 1The characteristics of PAAD.SampleaFeed ratio (wt%)Intrinsic viscosityViscosity-average molecular weightAMAADBDAPPAAD59.939.90.2772.12mL/g3.26×106aThe intrinsic viscosity and viscosity-average molecular weight were determined according to Refs.[28,29].where E is the total oil recovery ratio,E W is the oil recovery of water flooding.3.Results and discussion3.1.Characteristics of PAADThe effect of synthesis conditions on copolymerization of AM,AA and DBDAP were investigated,and the intrinsic viscosity and the viscosity-average molecular weight were measured.The results are summarized in Table 1(see Tables S1and S2and Fig.S1for the details in Supporting information).3.2.FTIR and 1H NMR spectra analysisFTIR and 1H NMR spectra of copolymer PAAD are shown in Fig.1,respectively.From the FTIR curve of PAAD,the strong absorp-tion peaks at 3434cm −1and 1651cm −1respectively assign tothe stretching vibration of N H and C O bond in the CONH 2group.A relatively less intense peaks at 1560and 1401cm −1are due to the COO −group [30].The peaks at 1325and 1119cm −1correspond to the stretching vibrations of SO 2.From the 1H NMR spectra of PAAD,the characteristic peaks around 1.54and 2.15ppm assign to the protons of polymer alkyl chains.The chem-ical shifts at 7.68and 6.87ppm are due to the protons of aromatic ring of DBDAP.It can be inferred that the typical structures of monomers have been successfully incorporated into polymer chain.3.3.Critical association concentration of PAADThe ratio of the intensities between the first and the third band intensity in the fluorescence spectrum of pyrene (I 1/I 3)is used to characterize the size of their environment polar-ity.The weaker polarity of the microenvironment around the pyrene molecule leads to the smaller value of I 1/I 3.Fig.2(a)depicts the relationship curve between the values of I 1/I 3and PAAD concentrations.On the curve of I 1/I 3,the value of I 1/I 3abruptly decreases at a concentration of PAAD about 1.5g/L suggesting the transformation of association type from intra-molecular association into intermolecular association.This is also evident from the curve of the viscosity versus concentration shown in Fig.2(b),and this value is defined as the criti-cal association concentration (CAC)at which the intramolecular association begins to transfer into intermolecular association [31].Fig.1.FTIR and 1H NMR spectra of PAAD.I 1 / I 3Concen tration (mg/L )(a)Concentration (mg/L)A p p a r e n t V i s c o s i t y (m P a ·s )(b)Fig.2.(a)Effect of PAAD concentration on I 1/I 3value;(b)effect of PAAD concentration on viscosity.S.Gou et al./Colloids and Surfaces A:Physicochem.Eng.Aspects471(2015)45–5349Fig. 3.Characteristics of different complex solutions(a)PAAD/C6mimBr;(b) PAAD/C8mimBr;(c)PAAD/C10mimBr;(d)PAAD/C12mimBr;(e)PAAD/C14mimBr;(f) PAAD/C16mimBr and(g)PAAD/CTAB.3.4.Characteristics of PAAD/ILs complex solutionsThe complex solutions of polymer PAAD and different ILs were obtained,and the complex of PAAD and CTAB was also pre-pared for comparing.CTAB is one of the most common used cationic surfactants for EOR.The concentration of PAAD wasfixed at3g/L.The photographs in Fig.3(a–g)show the different com-plex solutions,viz.,(a):PAAD/C6mimBr,(b):PAAD/C8mimBr,(c): PAAD/C10mimBr,(d):PAAD/C12mimBr,(e):PAAD/C14mimBr,(f): PAAD/C16mimBr and(g):PAAD/CTAB.However,when the concen-tration of C n mimBr,n=10,12,14and16,and CTAB is as low as 0.3g/L,a gel phase is observed in our experiment.This unexpected result can be owing to the strong binding of ion-pair interaction between cationic head groups and anion polymer corresponding to structural alkyl chain length of ILs.When the concentration of C n mimBr,n=6and8,is above40g/L,the solutions have remained transparent.The air/water critical micelle concentration(cmc)values of C6mimBr and C8mimBr were measured and compared with the values reported in the literatures at303.15K.The obtained cmc values of465mM for C6mimBr and118mM for C8mimBr are in agreement with the reported values470and121mM,respectively [32,33].Therefore the ILs concentrations werefixed at their cmcs in pure water to investigate the effect of ILs on complexes viscosity in the concentration of copolymer from1to5g/L.From Fig.3it is observed that with the concentration of C6mimBr around470mM, the viscosity of PAAD/C6mimBr complex decreases notably,e.g.the viscosity of3g/L PAAD decreases from660.5to8.7mPa s due to the large concentration of C6mimBr similarly to C8mimBr as discussed below.Because of the limitations of low viscosity of PAAD/C6mimBr complex and the high concentration of C6mimBr,studies of the interaction of IL and PAAD are focused on PAAD/C8mimBr complex.3.5.Effect of C8mimBr concentration on viscosityEffect of C8mimBr concentration on viscosity of2g/L and3g/L PAAD solutions is displayed in Fig.4.The concentration spanning a range from below to above the cmc of C8mimBr is actually higher than the39.9wt%anionic acrylic linked in PAAD chains at this mass ratio causing C8mimBr to partly incorporate with the PAAD and partly remain in the solutions which is also indicated in the TG-DSC results discussed below in this paper.Addition of C8mimBr causes an obvious decrease in the viscosity of complex solution due to the ionic C8mimBr reduces the electrostatic repulsion of polymerConcentration of IL (g/L)ApparentViscosity(mPa·s)Fig.4.Effect of C8mimBr concentration on complex viscosity: PAAD:3g/L,᭹PAAD:2g/L.chains,and cationic hydrophobic head groups adsorb on the anionic PAAD by opposite ion charge interaction leading to the polymer coils much more compact.The hydrophobic effect is apparently too strong in this system for the polymer coils to expand[8].3.6.Interfacial tension test3.6.1.Effect of C8mimBr concentration on interfacial tensionThe IFT changes versus PAAD/C8mimBr complex and C8mimBr at different concentrations of C8mimBr are depicted in Fig.5(a).The concentration of PAAD wasfixed at3g/L.The amphiphilic C8mimBr tends to migrate the interface leading to the adsorption,and con-sequently dropping the IFT.The lower values of IFT of C8mimBr are relevant in the presence of hydrophobically associating copolymer PAAD.For instance,the IFT decreases from2.1mN/m corresponding to30g/L C8mimBr to a minimum value of0.77mN/m correspond-ing to30g/L C8mimBr combined with3g/L PAAD.Accordingly,we carried out the study and discuss below on PAAD/C8mimBr with the30g/L C8mimBr.This can be explained by the surface activity of the copolymer and the Na+originated from PAAD copolymer solution.In details,adsorption of copolymer at the surface would necessarily compress the area available for ILs adsorption leading to the increase of surface excess ILs concentration and causing a lowering of the interface tension[8].In addition,Na+has higher surface charge density.The stronger hydration will be,the smaller number of water molecules available to hydrate[C8mim]+as a result of salting out effect[34].To further increase the concentra-tion of C8mimBr in PAAD solution,the higher surface tension of PAAD/C8mimBr complex demonstrates binding of ILs to the copoly-mer and concomitant depletion of ILs from the interface.3.6.2.Effect of polymer concentration on interfacial tensionAs presented in Fig.5(b),the effect of PAAD concentration from 1to5g/L on IFT of PAAD/C8mimBr complex and crude oil is inves-tigated.The results demonstrate a higher concentration above the CAC of PAAD did not modify the IFT significantly.The changes of concentration of polymer have no obvious effect on IFT of system due to C8mimBr forms mixed micelle with hydrophobic groups attached to the polymer.The concentration of PAAD/C8mimBr com-plex in the following research is3g/L PAAD with30g/L C8mimBr unless noted.3.6.3.Effect of temperature on interfacial tensionIn this stage,the effect of temperature(303K,308K,313K, 323K,333K and338K)on the IFT of PAAD/C8mimBr solution and50S.Gou et al./Colloids and Surfaces A:Physicochem.Eng.Aspects 471(2015)45–53I n t e r f a c i a l T e n s i o n (m N /m )Concentration of C 8mimBr (g/L)(a)I n t e r f a c i a l T e n s i o n(m N /m )Concentration of PAAD (g/L)(b)I n t e r f a c i a l T e n s i o n (m N /m )Temperure (oC)(c)I n t e r f a c i a l T e n s i o n (m N /m )Concentration of NaCl (g/L)(d)Fig.5.(a)Effect of C 8mimBr concentration on IFT;(b)effect of PAAD concentration on IFT;(c)effect of temperature on interfacial tension;(d)effect of NaCl concentration on interfacial tension: C 8mimBr:3wt%,᭹C 8mimBr:1wt%, C 8mimBr:0.5wt%, C 8mimBr:0.2wt%.crude oil was studied,and the effect of temperature on the IFT of C 8mimBr and crude oil was also investigated.The obtained results are given in Fig.5(c).The increasing temperature leads to the increase of IFT between C 8mimBr solutions and crude oil.This is because of the presence of nitrogen atoms with sp2hybridization in and the positive charge is in resonance,thus the diffusion of ILs into the oil phase increases as temperature increases leading to emulsion inversion from oil-in-water to water-in-oil resulting in the increase of IFT [20].However,the results reveal that with the temperature increase the IFT of PAAD/C 8mimBr solution and crude oil slightly decreases,which may be due to the hydrophobic asso-ciation of PAAD enhances as the temperature increases within a certain scope leading to lower IFT values.3.6.4.Effect of NaCl concentration on interfacial tensionThe effect of NaCl concentration on the IFT of PAAD/C 8mimBr solution and crude oil with different concentrations of C 8mimBr was examined.The results given in Fig.5(d)revealed that the IFT reduced at higher NaCl concentrations due to the enhancement of hydrophobic association of complex and the salting out effect discussed earlier in this paper.For instance,the complex of 3g/L PAAD and 5g/L C 8mimBr with 20g/L NaCl can reduce the IFT to 0.85mM/m.In a word,with electrolytes,the interaction param-eter tends to higher positive values indicating reduction in the repulsive interactions between cationic head group of IL molecules [34].This observed trend makes the PAAD/C 8mimBr complex aseffective alternative for EOR processes dealing with harsh salinity conditions.3.7.Effect of temperature and shear rate on viscosityThe effect of temperature on the apparent viscosity of PAAD (2g/L)and PAAD/C 8mimBr complex solutions at a shear rate of 170s −1is shown in Fig.6(a).The viscosity of PAAD decreases then increases followed by a decrease,and it attains a maximum value at 90◦C.This may be due to that the high temperature can enhance hydrophobic association of PAAD.However,to further increase the temperature,the hydrophobic groups are disrupted,so that the viscosity decreased.The viscosity of PAAD/C 8mimBr complex maintains slight decrease from 22.2to 17.4mPa s with temperature raising from 30to 90◦C showing excellent temperature resistance.This decrease may be due to the more enhancing hydrophobic effect with the increasing temperature in this system limiting the poly-mer coils to expand.The shear thinning behavior of PAAD (2g/L)and PAAD/C 8mimBr complex solutions was measured,and the results are shown in Fig.6(b).The shear thinning behavior and reversible are important for polymer injection.At high shear rate,the apparent viscosity of PAAD and PAAD/C 8mimBr complex solutions exhibits a significant decrease.To research the recoverability to alteration in the shear rate,the sample solutions maintained shearing at 170s −1for 5min,next kept shearing at 500s −1for 5min,then went on shearing atS.Gou et al./Colloids and Surfaces A:Physicochem.Eng.Aspects 471(2015)45–535110100A p p a r e n t V i s c o s i t y (m P a ·s )Temperature (oC)(a)Shear Rate (s-1)A p p a r e n t V i s c o s i t y (m P a ·s )(b)Shear Rate (s -1)A p p a r e n t V i s c o s i t y (m P a ·s )Time (s)(c)100200300400500Shear Rate (s -1)A p p a r e n t V i s c o s i t y (m P a ·s )Time (s)(d)100200300400500Fig.6.(a)Effect of temperature on viscosity;(b)effect of shear rate on viscosity;(c)recovering ability of PAAD for shear rate;(d)recovering ability of PAAD/C 8mimBr for shear rate.170s −1for 5min.The results are shown in Fig.6(c,d).When shear rate suddenly changes from 170to 500s −1,the viscosity of PAAD and PAAD/C 8mimBr drops sharply,and when shear rate decreases from 500to 170s −1,the viscosity of PAAD and PAAD/C 8mimBr com-plex recovers immediately.About 87.6%viscosity retention rate compared with the original viscosity is obtained by PAAD,and for PAAD/C 8mimBr complex,the viscosity is equal to the original vis-cosity.It has been shown that the interaction between PAAD and C 8mimBr has excellent recovering ability for shear rate.3.8.TG and DSCTG and DSC were used to analyze the thermal decomposition of PAAD and PAAD/C 8mimBr complex.The results are presented in Fig.7(a,b).As shown in TG diagram of PAAD,the thermogravi-metric stage occurs with the mass loss of 77.96wt%which could be attributed to the decompositions and carbonization of copoly-mer.The TG diagram of PAAD/C 8mimBr displays two stages for the weight loss.The first step occurs in the range of 40–450◦CW e i g h (%)Temperture (oC)(a)D S C (m W /m g )Temperture (oC)(b)Fig.7.(a)TG diagram of PAAD and PAAD/C 8mimBr;(b)DSC diagram of PAAD and PAAD/C 8mimBr.。

ElsevierAccounts of Chemical Research 1968- Acta Materialia 1997-Additives for Polymers 1997- Advanced Drug Delivery Reviews 1997-Advances in Colloid and Interface Science 1995-Advances in Environmental Research 1997-Analytical Biochemistry 1997- Analytica Chimica Acta 1995-Antiviral Research 1997- Applied Catalysis A: General 1997-Applied Catalysis B: Environmental 1997- Applied Clay Science 1997-Applied Surface Science 1995- Archives of Biochemistry and Biophysics 1997- Biochemical and Biophysical Research Communications 1997-Biochemical Engineering Journal 1997- Biochemical Pharmacology 1997-Biochemical Systematics and Ecology 1997- Biochimie 1997-Bioelectrochemistry 2000- Bioelectrochemistry and Bioenergetics 1995-1999Biomaterials 1997- Bioorganic Chemistry 1995-Bioorganic & Medicinal Chemistry 1995- Bioorganic & Medicinal Chemistry Letters Biophysical Chemistry 1995- Biosensors and Bioelectronics 1995-Biotechnology Advances 1997- Carbohydrate Research 1995-Catalysis Communications 1997- Catalysis Today 1997-Chemical Engineering Science 1997- Chemical Health and Safety 1995-Chemical Physics 1995- Chemical Physics Letters 1995-Chemistry & Biology 1995- Chemistry and Physics of Lipids 1997-Chemometrics and Intelligent Laboratory Systems 1998- Chemosphere 1997-Colloids and Surfaces A: Physicochemical and Engineering Aspects 1995-Colloids and Surfaces B: Biointerfaces 1995-Combinatorial Chemistry - an Online Journal 2003-Comptes Rendus de l'Académie des Sciences - Series IIB - Mechanics-Physics-Chemistry-Astronomy 1995-Comptes Rendus de l'Académie des Sciences - Series IIC - Chemistry 1998-2001 Comptes Rendus Chimie 2002-Computational and Theoretical Polymer Science 1997-2001Computational Biology and Chemistry 2003- Computers & Chemistry 1995-2002 Coordination Chemistry Reviews 1995-Critical Reviews in Analytical Chemistry 2003-Crystal Engineering 1999- Current Biology 1997-Current Opinion in Biotechnology 1997- Current Opinion in Cell Biology 1997-Current Opinion in Chemical Biology 1997-Current Opinion in Colloid & Interface Science 1999-Current Opinion in Genetics & Development 1997-Current Opinion in Pharmacology 1997- Current Opinion in Plant Biology 1997- Current Opinion in Structural Biology 1997-Drug Discovery Today 1997- Dyes and Pigments 1997-Electrochemistry Communications 1999-Electrochimica Acta 1995- Environmental Toxicology and Pharmacology 1997- Enzyme and Microbial Technology 1997-European Journal of Medicinal Chemistry 1995-European Journal of Pharmaceutical Sciences 1997-European Journal of Pharmaceutics and Biopharmaceutics 1997-European Journal of Pharmacology 1997-European Journal of Solid State and Inorganic Chemistry 1998European Polymer Journal 1995-Experimental Cell Research 1997- Experimental Neurology 1997-FEBS Letters 1997- FEMS Microbiology Letters 1997-Fluid Phase Equilibria 1995- Focus on Polyvinyl Chloride 2002Focus on Powder Coatings 2002- Forensic Science International 1995-Free Radical Biology and Medicine 1997-Fuel 1995- Fuel Processing Technology 1995-Gene 1997- Inorganic Chemistry Communications 1995-Inorganica Chimica Acta 1995-International Journal of Adhesion and Adhesives 1997-International Journal of Antimicrobial Agents 1997-International Journal of Biological Macromolecules 1997-International Journal of Hydrogen Energy 1995-International Journal of Inorganic Materials 1999-2001International Journal of Mass Spectrometry 1998-International Journal of Mass Spectrometry and Ion Processes 1995-1998International Journal of Pharmaceutics 1997-International Journal of Biochemistry and Cell Biology 1998-Journal of Alloys and Compounds 1997-Journal of the American College of Cardiology 1997-Journal of the American Society for Mass Spectrometry 1995-Journal of Analytical and Applied Pyrolysis 1995-Journal of Biochemical and Biophysical Methods 1997-Journal of Bioscience and Bioengineering 1997-Journal of Biotechnology 1997- Journal of Catalysis 1997-The Journal of Chemical Thermodynamics 1995-Journal of Chromatography A 1995-Journal of Chromatography B: Analytical T echnologies in the Biomedical and Life Sciences 2002-Journal of Chromatography B: Biomedical Sciences and Applications 1995-2001Journal of Colloid and Interface Science 1995-Journal of Controlled Release 1997- Journal of Cultural Heritage 1997-Journal of Electroanalytical Chemistry 1995-Journal of Electron Spectroscopy and Related Phenomena 1995-Journal of Fluorine Chemistry 1995-Journal of Food Composition and Analysis 1997-Journal of Inorganic Biochemistry 1995-Journal of Luminescence 1995- Journal of Magnetic Resonance 1997-Journal of Membrane Science 1997- Journal of Microbiological Methods 1997-Journal of Molecular Biology 1997- Journal of Molecular Catalysis A: Chemical 1997- Journal of Molecular Catalysis B: Enzymatic 1997-Journal of Molecular and Cellular Cardiology 1997-Journal of Molecular Graphics 1995-1996Journal of Molecular Graphics and Modelling 1997-Journal of Molecular Liquids 1995-Journal of Molecular Spectroscopy 1995-Journal of Molecular Structure 1995-Journal of Molecular Structure: THEOCHEM 1995-Journal of Organometallic Chemistry 1995-Journal of Pharmaceutical and Biomedical Analysis 1995-Journal of Pharmacological and Toxicological Methods 1997-Journal of Photochemistry and Photobiology A: Chemistry 1995-Journal of Photochemistry and Photobiology B: Biology 1995-Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2000-Journal of Power Sources 1995-Journal of Quantitative Spectroscopy and Radiative Transfer 1995-Journal of Solid State Chemistry 1995-Journal of Structural Biology 1997-The Journal of Supercritical Fluids 1995-Journal of Supramolecular Chemistry 2001-2002Laboratory Automation & Information Management 1998-1999Marine Chemistry 1995- Materials Chemistry and Physics 1997-Materials Letters 1997- Materials Science and Engineering A 1997- Materials Science and Engineering B 1997-Materials Science and Engineering C 1995-Materials Science and Engineering R: Reports 1997-Materials Today 1997- Microchemical Journal 1995-Microporous and Mesoporous Materials 1997-Molecular and Cellular Neuroscience 1997-Molecular Immunology 1997- Neurochemistry International 1997- Neuroscience Letters 1997- Optical Materials 1997-Organic Electronics 1997- Peptides 1997-Pesticide Biochemistry and Physiology 1997-Pharmaceutical Science & Technology Today 1997-Pharmacological Research 1997- Pharmacology & Therapeutics 1997- Phytochemistry 1995- Plasmas & Ions 1998-2000Polyhedron 1995- Polymer 1995- Polymer Contents 1995-Polymer Degradation and Stability 1995- Polymer Testing 1995-Powder Technology 1995- Progress in Lipid Research 1997-Progress in Nuclear Magnetic Resonance Spectroscopy 1995-Progress in Organic Coatings 1995-Progress in Polymer Science 1995-Progress in Surface Science 1995-Protein Expression and Purification 1995-Reactive and Functional Polymers 1995-Revue Franaise des Laboratoires 1999-Sensors and Actuators A: Physical 1995-Sensors and Actuators B: Chemical 1995-Separation and Purification Technology 1997-Solar Energy Materials & Solar Cells 1992-Solid State Ionics 1995- Solid State Sciences 1999-Solid State Nuclear Magnetic Resonance 1995-Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 1997- Spectrochimica Acta Part A: Molecular Spectroscopy 1995-1996 Spectrochimica Acta Part B: Atomic Spectroscopy 1995-Steriods 1995- Structure 1997-Supramolecular Science 1995-1998Surface and Coatings Technology 1997-Surface Science 1995-Surface Science Reports 1995-Synthetic Metals 1997- Talanta 1995-Tetrahedron 1995- Tetrahedron: Asymmetry 1995-Tetrahedron Letters 1995- Thermochimica Acta 1995-Thin Solid Films 1997- Toxicological Sciences 1997-Toxicology 1997- Toxicology and Applied Pharmacology 1997-Toxicology in Vitro 1997- Toxicology Letters 1997-Toxicon 1997- Trends in Biochemical Sciences 1997-Trends in Biotechnology 1997- Trends in Cell Biology 1997-Trends in Molecular Medicine 1997- Trends in Pharmacological Sciences 1997- Vibrational Spectroscopy 1995-。


Available online at Colloids and Surfaces A:Physicochem.Eng.Aspects312 (2008) 99–103In situ and one-step synthesis of hydrophobic zinc borate nanoplatelets Yumei Tian,Yi He,Lianxiang Yu,Yanhui Deng,Yunhui Zheng,Fang Sun,Zhihui Liu,Zichen Wang∗College of Chemistry,Jilin University,Changchun130012,ChinaReceived22March2007;received in revised form31May2007;accepted14June2007Available online 19 June 2007AbstractThe polycrystalline and hydrophobic zinc borate(Zn2B6O11·3H2O)nanoplatelets were in situ successfully synthesized via one-step precipitation reaction in aqueous solution of Na2B4O7·10H2O and ZnSO4·7H2O with oleic acid as the modifying agent.The microstructures and morphology of the as-obtained samples were studied by X-ray diffraction(XRD),scanning electron microscopy(SEM)equipped with an energy-dispersive X-ray spectrometer(EDS),transmission electron microscopy(TEM)and thermogravimetric analysis(TGA).Measurements of the relative water contact angle and the active ratio indicated that Zn2B6O11·3H2O samples were hydrophobic.It had been found that the as-prepared materials displayed nanoplatelet morphology with average diameters100–500nm and thickness30±5nm and the morphology and size of the samples were controlled effectively.© 2007 Elsevier B.V. All rights reserved.Keywords:Zinc borate;Nanoplatelets;Hydrophobe;Active ratio;Synthesis1.IntroductionZinc borate has been the subject of significant research for applications including the polymer additive which serves as the char promoter,theflame retardant synergist,the preservative in wood composites,the smoke and afterglow suppressant due to its ability to undergo endothermic dehydration infire con-ditions[1–4],and optical properties[5–8],and the additive for lubrication[9].Previous work about the synthesis of zinc borate included by reactions of zinc salts and borate salts in hot water(≥60◦C)or through the ethanol supercriticalfluid drying technique[10],and reaction of zinc oxide and boric acid[11].Important attributes of zinc borate include relatively low water solubility and a relatively high dehydration onset temperature[12].The latter property permits processing in a wide range of polymer system.But zinc borate particles are hardly dispersed in a polymer matrix so that they prevent their using in industry.To the best of our knowledge,studies of the preparation of zinc borate with nanos-tructures and the hydrophobic properties have been relatively few.Herein we report the in situ and one-step synthesis of active ∗Corresponding author.Tel.:+8643185155358;fax:+8643188499134.E-mail address:wangzc@(Z.Wang).zinc borate nanoplatelets,which requires neither sophisticatedtechniques nor catalysts.Surface modification of zinc boratewith hydrophobic properties would lead to a great expansion ofits applications.2.Experimental part2.1.MaterialsAll reagents purchased from Beijing Chemicals Co.Ltd.wereof analytical purity and employed without any further treat-ments.Distilled water was applied for all synthesis and treatmentprocesses.2.2.Synthesis of samplesIn a typical procedure,a500mL three-neck round-bottomedflask equipped with a thermometer,reflux condenser,andmechanical stirrer was charged with100mL of0.1mol dm−3Na2B4O7·10H2O aqueous solution,20.0mL of absolute ethanol and a certain amount of oleic acid(OA)heated at70◦C and10.0mL of2mol dm−3ZnSO4·7H2O aqueous solution was added dropwise to thefirst solution while being stirred for a period of about0.5h.After addition was complete,the mixture0927-7757/$–see front matter© 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.colsurfa.2007.06.029100Y.Tian et al./Colloids and Surfaces A:Physicochem.Eng.Aspects 312 (2008) 99–103(pH<7.0)was continuously heated for about6.5h.Thefinalmixture was thenfiltered,washed repeatedly with absoluteethanol and distilled water to remove unreacted reactants andby-products,and dried in the oven at80◦C to obtain thefinalwhite zinc borate powders.2.3.Characterization of the samplesThe structure and composition of as-prepared composites wascharacterized by X-ray power diffraction(XRD)(SHIMADZUXRD-6000diffractometer employing Ni-filtered Cu K␣radi-ation,at a scanning rate of6◦/min with2θranging from20◦to45◦)and an energy-dispersive X-ray spectrometer(EDS)(JEOL-6700F,Hitachi)attached to the SEM,respectively.The morphology and size of the samples were observed usinga Hitachi H-800transmission electron microscope(TEM),atan accelerator voltage of200kV and a Hitachi scanning elec-tron microscope(SEM)with afield-emission-scanning electronmicroscope(JEOL-6700F,20kV).The samples used for SEMand TEM characterization were dispersed in absolute ethanoland were ultrasonicated before observation.Thermogravimetric analysis(TGA)was carried out using aDTG-60H analyzer(SHIMAPZU).Tests were performed withabout10mg of sample on aluminum crucibles with the heatingrates of10◦C/min from50to500◦C in air atmosphere and usingtabular␣-Al2O3as reference weight losses were calculated fromthe TGA.The effect of surface modification is evaluated by the rela-tive water contact angle andfloating test.Water contact angle ofsample was measured by using a FT˚A200(USA)contact angleanalyzer.Onflat solid surfaces,the water contact angle can becarried out through the sessile drop technique.The powder sam-ple wasfirst pressed into a wafer under10MPa pressure andthen was measured by sessile drop method.This method offersa two-fold advantage,that is,it requires a very small amount ofliquid and a scant solid surface.Thefloating test was used tomeasure the ratio of thefloating product to the overall weight ofthe sample after they were mixed in water and stirred vigorously[13].3.Results and discussion3.1.Structural and morphological characteristics of zinc borateThe synthesized product is stable white powder at room tem-perature.Fig.1shows the XRD patterns of the products.Somesharp peaks of the pure zinc borate were shown in Fig.1a,whichindicated the particles were crystals.All diffraction peaks werequite similar to those of bulk Zn2B6O11·3H2O and the diffrac-tion data were in good agreement with JCPDSfile No.21-1473and earlier reports[10].No characteristic peaks of impuri-ties,such as Na2SO4,other zinc borates and other unreactedcompounds were observed.Thus,the results showed that the as-prepared product was polycrystalline phase Zn2B6O11·3H2O. The XRD of hydrophobic zinc borate resembled that of pure zinc borate(shown in Fig.1b).Chemical analysis using EDStaken Fig. 1.XRD of(a)pure Zn2B6O11·3H2O powders and(b)hydrophobic Zn2B6O11·3H2O nanoplatelets.from the as-prepared pure zinc borate particles(Fig.4a)indi-cated the presence of Zn,B and O(elements of Zn2B6O11·3H2O and element H was not analyzed),no other such as S existed in Fig.2.Furthermore,the quantification of the peaks gave that the atomic ratio of Zn:B:O was2:6.03:14.12,which was very close to the stoichiometric Zn2B6O11·3H2O.Typical SEM images and TEM patterns of the products are shown in Figs.3and4.Fig.3shows SEM patterns of nanocrys-tals made with different zinc sulfate/sodium borate mole ratios. 2:1zinc sulfate/sodium borate and without OA added gave poly-hedral shape zinc borate(Fig.3a).1:1zinc sulfate/sodium borate mole ratio and1.00wt%of OA added also gave polyhedral shape zinc borate(Fig.3b).Increasing the zinc sulfate/sodium borate mole ratio to1.5:1(1.00wt%of OA added)gave only zinc borate nanoplatelets(Fig.3c)and increasing the zinc sul-fate/sodium borate ratio to2:1(1.00wt%of OA added)also gave zinc borate nanoplatelets(Fig.3d).The diameters of the nanoplatelets were from100to500nm and the thicknesses of the nanoplatelets could be estimated to be30±5nm.It could also be found that the TEM pattern of the zinc borate without sur-factant(see Fig.4a)showed polyhedral shape with the particle size of around50–500nm,while hydrophobic zinc borate(see Fig.4b)showed platelet-like morphology.It was evident that the morphologies transformed from irregular to regular,indicat-ing that OA and different reactants mole ratios could effectively adjust the shape of the particles.In addition,the SAED(select area electron diffraction)pattern(inset in Fig.4a)consisted ofaFig.2.Typical EDS spectrum of pure zinc borate sample.Y.Tian et al./Colloids and Surfaces A:Physicochem.Eng.Aspects 312 (2008) 99–103101Fig.3.SEM images of(a)pure zinc borate particles and modified zinc borate particles(1.00wt%of OA added)with different zinc sulfate/sodium borate mole ratios: (b)1:1,(c)1:1.5,and(d)1:2.number of rings,suggesting a polycrystalline structure,consis-tent with those data of XRD diffraction of the pure zinc borate powders(Fig.1a).But the SAED(inset in Fig.4b)was taken from the obtained hydrophobic zinc borate nanoplatelets,which diffraction“rings”consisted of some distinct spots along the ring contours.The rings in an electron diffraction pattern arised due to the diffracted electron beam from a set of lattice planes in the crystallites present in the sample satisfying the Bragg diffrac-tion condition.In other words,the ring was an envelope of all diffracted spots associated with nanoclusters formation[14].3.2.TGA of the composite nanoplateletsIn order to confirm the oleic acid bonded onto the surface of zinc borate,samples were analyzed by TGA.Fig.5repre-sents TGA plots of the pure zinc borate,hydrophobic zincborate Fig.4.TEM images of(a)pure zinc borate particles and(b)obtained hydrophobic zinc borate nanoplatelets(insets are SAED patterns).102Y.Tian et al./Colloids and Surfaces A:Physicochem.Eng.Aspects312 (2008) 99–103Fig. 5.TGA of(a)the pure Zn2B6O11·3H2O powders,(b)hydrophobic Zn2B6O11·3H2O nanoplatelets,and(c)pure OA.(1.00wt%of OA added)and OA.For the pure zinc borate(shown in Fig.5a),there was the total weight loss of14.201wt%when the sample was heated from120to500◦C.Fig.5a curve indi-cated that the weight loss was12.793%from120to440◦C, which corresponded to the loss of three molar equivalents of the crystal water and could be compared with calculated value of 12.697%and formed2ZnO·3B2O3.Beyond440◦C,the weight changed very little,and the weight loss in this temperature range was due to the decomposition of hydroxide ly, there were hydroxide groups in the surface of zinc borate par-ticles.OA began to decompose from130◦C and decomposed completely at440◦C(Fig.5c).The hydrophobic zinc borate sample(Fig.5b),into which1.00wt%of OA was added,had a total weight loss of15.202wt%from120to500◦C.Weight loss increased quickly from190to320◦C,because of the decompo-sition of the organic groups of OA and the compound containing the crystal water.The amount of OA calculated by TGA equaled to that added in the preparation process of raw materials.3.3.Surface property of zinc borate nanoplateletsThe contact water angle is widely used as a criterion for evaluating surface hydrophobicity.In order to study the surface characteristics,the zinc borate powder is analyzed with measur-ing the relative contact angle and the active ratio by thefloating test,respectively.Fig.6presented a plot of the active ratio ver-sus the amount of the OA.The active ratio increased quickly from60.1to99.9%with weight ratio of OA/Zn2B6O11·3H2O increasing from0.5to1.0wt%.The fact that the obtained sam-ple’s active ratio was effectively demonstrated that theorganic Fig.6.Influence of the amount of oleic acid on the active ratio of obtained Zn2B6O11·3H2O powders.group of OA had been possibly adsorbed onto the surface of Zn2B6O11·3H2O and the optimal amount of OA was1.0wt%.The contact angle was often used to measure the extent of hydrophobicity of solid surfaces.Onflat solid surfaces,it is easy that the contact angle can be measured by the ses-sile drop technique with commercially available instruments. The images of synthesized Zn2B6O11·3H2O powder using var-ious amount of oleic acid by the water drop shape technique were given in Fig.7.It can be seen that the water droplet dropped on pure Zn2B6O11·3H2O powder of wafer(Fig.7a) did not form the changeless drop shape,but was imbibed by pure Zn2B6O11·3H2O powder.This phenomenon suggested that the contact angle of pure Zn2B6O11·3H2O powder was very little and hydrophilic.So that Zn2B6O11·3H2O was very easy to be wetted by water and the relative contact angle was71◦. When the oleic acid(0.5wt%)was added into the process of the preparation of Zn2B6O11·3H2O,its wettability was decreased, and the relative contact angle was increased to116◦(Fig.7b). When the amount of oleic acid was increased to1.0wt%,the droplet placed on it remained spherical and the relative contact angle was increased to132◦(Fig.7c),which induced a change of the Zn2B6O11·3H2O powders surface from hydrophilic to hydrophobic.The data of relative contact angles consisted with the analysis of the active ratio.At the same time,the pH val-ues of the reaction solution also showed a significant effect on the hydrophobicity of Zn2B6O11·3H2O powder in the presence of OA.Fig.7also shows the contact angles of Zn2B6O11·3H2O obtained at different pH values adjusted by using2mol/L NaOH or HCl.The relative contact angle was decreased to101◦at the pH7of the solution(Fig.7d),and the active ratio alsoreducedFig.7.Behavior of water droplet on thin pellet of Zn2B6O11·3H2O nanoparticles surface of images with various amount of OA and different pH values:(a)0wt%, pH5.8;(b)0.5wt%,pH5.8;(c)1.0wt%,pH5.8;(d)1.0wt%,pH7.Y.Tian et al./Colloids and Surfaces A:Physicochem.Eng.Aspects 312 (2008) 99–103103Fig.8.Influence of pH on the active ratio of obtained Zn2B6O11·3H2O powders. greatly.When pH is7.0and above7.0the active ratio of zinc borate powder is lower(shown in Fig.8).The active ratio can reach100%only if pH was lower than7.0and the optimum pH was5.6–6.0(shown in Figs.7and8).When the solution is in the weak acid medium,the surface of zinc borate particles had more positive charges so that the anionic surfactant OA could be easily absorbed into the surface of zinc borate.From above discussed, we concluded that the pH value of the solution greatly influenced the relative contact angle and active ratio of zinc borate.Oleic acid was bonded to the O–H groups at the surface of zinc borate with a single hydrogen bond[9].4.ConclusionsHydrophobic Zn2B6O11·3H2O nanoplatelets were in situ successfully obtained by Na2B4O7·10H2O and ZnSO4·7H2O as raw materials through one-step precipitation reaction,and OA as the modifying agent.The optimal amount of OA was 1.0%of the weight of Zn2B6O11·3H2O and the optimum pH was5.6–6.0.XRD analysis indicated that the product was high purity and polycrystalline phase of Zn2B6O11·3H2O.SEM anal-ysis indicated that when the zinc sulfate/sodium borate mole ratio to1.5:1–2:1(1.00wt%of OA added)gave the morphol-ogy of product only displayed platelet morphology with average diameters100–500nm and thicknesses30±5nm.Results anal-ysis indicated the oleic acid was bonded onto the surface of Zn2B6O11·3H2O with a single hydrogen bond.At the same time, the measurement of the relative contact angle and the active ratio indicated that Zn2B6O11·3H2O samples were hydrophobic.References[1]H.E.Eltepe,D.Balk¨o se,S.¨Ulk¨u,Effect of temperature and time on zincborate species formed from zinc oxide and boric acid in aqueous medium, Ind.Eng.Chem.Res.46(2007)2367.[2]X.Chen,Y.Zhao,X.Chang,J.Zuo,H.Zang,W.Xiao,Syntheses andcrystal structures of two new hydrated borates,Zn8[(BO3)3O2(OH)3]and Pb[B5O8(OH)]·1.5H2O,J.Solid State Chem.179(2006)3911.[3]S.Bourbigot,M.L.Bras,R.Leeuwendal,K.K.Shen,D.Schubert,Recentadvances in the use of zinc borates inflame retardancy of EV A,Polym.Degrad.Stabil.64(1999)419.[4]F.Carpentier,S.Bourbigot,M.L.Bras,R.Delobel,M.Foulon,Charring offire retarded ethylene vinyl acetate copolymer-magnesium hydroxide/zinc borate formulations,Polym.Degrad.Stabil.69(2000)83.[5]S.E.Dann,M.T.Weller,B.D.Rainford,D.T.Adroja,Synthesis,struc-ture,optical properties,and magnetism of the manganese chalcogenide beryllosilicate and beryllogermanate sodalites,Inorg.Chem.36(1997) 5278.[6]U.Visk,A.Suisalu,J.Kikas,A.Osvet,A.Winnacker,Spectral diffusionbroadening by soft local modes in Sm2+-doped sodium borate glass,J.Lumin.127(2007)19.[7]N.S.Hussain,N.Ali,A.G.Dias,M.A.Lopes,J.D.Santos,S.Buddhudu,Absorption and emission properties of Ho3+doped lead–zinc–borate glasses,Thin Solid Films515(2006)318.[8]A.Ivankov,J.Seekamp,W.Bauhofer,Optical properties of Eu3+-dopedzinc borate glasses,J.Lumin.12(2006)123.[9]Y.Tian,Y.Guo,M.Jiang,Y.Sheng,B.Hari,G.Zhang,Y.Jiang,B.Zhou,Y.Zhu,Z.Wang,Synthesis of hydrophobic zinc borate nanodiscs for lubrication,Mater.Lett.60(2006)2511.[10]J.X.Dong,Z.S.Hu,A study of the anti-wear and friction-reducing proper-ties of the lubricant additive,nanometer zinc borate,Tribol.Int.31(1998) 219.[11]A.V.Shete,S.B.Sawant,V.G.Pangarkar,Kinetics offluid-solid reactionwith an insoluble product:zinc borate by the reaction of boric acid and zinc oxide,J.Chem.Technol.Biotechnol.79(2004)526.[12]D.M.Schubert, F.Alam,M.Z.Visi, C.B.Knobler,Structural char-acterization and chemistry of the industrially important zinc borate Zn[B3O4(OH)3],Chem.Mater.15(2003)866.[13]L.Tang,B.Zhou,Y.Tian,H.Bala,Y.Pan,S.Ren,Y.Wang,X.Lv,M.Li,Z.Wang,Preparation and surface modification of uniform ZnO nanorods via a one-step process,Colloid Surf.A:Physicochem.Eng.A296(2007)92.[14]Q.Liao,R.Tannenbaum,Z.L.Wang,Synthesis of FeNi3alloyednanoparticles by hydrothermal reduction,J.Phys.Chem.B110(2006) 14262.。

Colloids and Surfaces A:Physicochem.Eng.Aspects 370 (2010) 28–34Contents lists available at ScienceDirectColloids and Surfaces A:Physicochemical andEngineeringAspectsj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /c o l s u r faPreparation of monodispersed polyelectrolyte microcapsules with high encapsulation efficiency by an electrospray techniqueYu Fukui,Tatsuo Maruyama ∗,Yuko Iwamatsu,Akihiro Fujii,Tsutomu Tanaka,Yoshikage Ohmukai,Hideto Matsuyama ∗∗Department of Chemical Science and Engineering,Kobe University,1-1Rokkodai,Nada-ku,Kobe 657-8501,Japana r t i c l e i n f o Article history:Received 29May 2010Received in revised form 11August 2010Accepted 14August 2010Available online 21 August 2010Keywords:Electrospray Microcapsule PolyelectrolyteHigh encapsulation efficiencya b s t r a c tThe preparation of polyelectrolyte microcapsules by electrospray was investigated.When a polyanionic or polycationic aqueous solution was electrosprayed into an aqueous solution containing a polyelectrolyte with the opposite charge,a spherical interface consisting of a polyelectrolyte complex was formed by electrostatic interaction to produce a microcapsule.Alginate/chitosan microcapsules (∼100m)were successfully produced with a narrow diameter distribution (coefficient of variation 4.4%).The diameters of microcapsules were controlled in the range of 80–230m by varying the operating conditions,such as feed rate,working voltage,the distance from needle-to-collector,needle diameter and polyelectrolyte concentrations.We also succeeded in the encapsulation of protein,dextran and a polymeric microsphere within the polyelectrolyte microcapsules with high encapsulation efficiencies (more than 99%).The study of yeast encapsulation reveals that the electrospray technique can encapsulate a physiologically active substrate in the polyelectrolyte microcapsule and maintain its activity.© 2010 Elsevier B.V. All rights reserved.1.IntroductionMicroencapsulation has been widely used in the agricultural,food,cosmetics,pharmaceutical and medical industries [1–3].Along with the emerging demand for new functional microcap-sules,the development of a new microencapsulation technique has been a significant target in the research field to achieve effi-cient encapsulation,a biocompatible microcapsule,a functional microcapsule or a controlled-release strategy [4–6].Emulsifica-tion,spray-drying and coacervation techniques are mostly used for encapsulating food ingredients,proteins,drugs,flavors and liv-ing cells.In the last decade,studies in microfluidics technology have provided a novel strategy for the preparation of microcapsules with narrow size-distributions [7–9].The electrospray technique,which is conventionally used as an ionization technique in mass spectroscopy,has a great potential for microcapsule preparation because of the simplicity of the apparatus,its high productivity and its easy setup.Although the electrospray technique can con-tinuously produce tiny droplets or nanofibers with ease,there has been uncertainty around the production of microcapsules.Bugarski et al.first reported microcapsule preparation using the electro-∗Corresponding author.Tel.:+81788036070;fax:+81788036070.∗∗Corresponding author.E-mail addresses:tmarutcm@crystal.kobe-u.ac.jp (T.Maruyama),matuyama@kobe-u.ac.jp (H.Matsuyama).spray technique and demonstrated the effective preparation of size-controlled microcapsules [10].They and other research groups subsequently reported encapsulation of viable living cells using calcium alginate and the electrospray technique [11–14].These reports employed only calcium alginate as a capsule material,prob-ably due to its safety with living cells.In 1980s,relatively large capsules (∼mm)composed of poly-electrolytes were studied to encapsulate cells and enzymes [15,16].These capsules of millimeter size were prepared simply by adding a charged-polyelectrolyte solution dropwise to another oppositely charged-polyelectrolyte solution.In the last decade,the layer-by-layer method using polyelectrolytes have attracted wide attention as a thin film material because they form a stable and insolu-ble complex with an oppositely charged polyelectrolyte [17].In particular,this method can employ synthetic and natural poly-electrolytes and provide a novel class of functional ultrathin films.The functional properties of polyelectrolyte films (e.g.,controlled release of encapsulated compounds,self-rupturing,biocompatibil-ity and stimuli-responsiveness,etc.)are generally derived from the design and combination of polyelectrolytes [18–21].In some cases,the solubility of the complex formed depends on the pH and the ionic strength of the solution,which was also used for the controlled release of encapsulated compounds or for the stimuli-responsiveness of the thin film.The layer-by-layer method has been also extended to prepare microcapsules using a sacrifice template [22,23].However,the use of a sacrifice template makes it difficult to effectively encapsulate core substrates within a microcapsule.0927-7757/$–see front matter © 2010 Elsevier B.V. All rights reserved.doi:10.1016/j.colsurfa.2010.08.039Y.Fukui et al./Colloids and Surfaces A:Physicochem.Eng.Aspects370 (2010) 28–3429The strong electrostatic interaction and fast complex-formation between positively and negatively charged polyelectrolytes require the manipulation of each aqueous solution with another immisci-ble phase(e.g.,a water-immiscible organic solvent or a gas phase) when preparing microcapsules composed of polyelectrolytes.The electrospray technique ejects a droplet to a gas phase at high speed and the droplet falls onto a collector vessel.We expected that the electrospray technique would enable the preparation of monodispersed microcapsules based on cationic and anionic polyelectrolytes with high encapsulation efficiency. The present study reports the preparation of microcapsules using several kinds of polyelectrolytes by the electrospray technique. Most of the experiments were performed using chitosan and alginate as polyelectrolytes because this polyelectrolyte combi-nation was widely applied for capsule production,as can be seen in the following references[24–26].We investigated key parameters of the electrospray technique to control the size of microcapsules and found that the electrospray technique could produce monodispersed microcapsules(around100m in diam-eter)and also encapsulate various materials(biomacromolecules, microparticles and living cells)within the microcapsules with high encapsulation efficiency.2.Experimental2.1.MaterialsSodium alginate,chitosan,acetic acid and sodium hydrox-ide were purchased from Wako Pure Chemical Industries(Osaka, Japan).Poly(sodium4-styrenesulfonate)(PSS,Mw=∼70,000),poly (allylamine hydrochloride)(PAH,Mw=∼56,000),albumin–fluore-scein isothiocyanate conjugate(albumin–FITC),and tetramethyl-rhodamine isothiocyanate–dextran(TRITC–dextran)with average molecular weights of4400,65,000–76,000and155,000were pur-chased from Sigma(St.Louis,MO).Poly(diallyldimethylammonium chloride)solution(PDDA,Mw=40,000,28wt%in H2O)was pur-chased from Polysciences Inc.(Warrington,PA).Greenfluorescent polystyrene microspheres were purchased as a suspension from Duke Scientific(Palo Alto,CA).The microspheres had a diame-ter of1.9m.Yeast extract and peptone were purchased from Becton,Dickinson and Company(Sparks,MD).d-(+)-Glucose was purchased from Nacalaitesque,Inc(Kyoto,Japan).2.2.ElectrosprayThe electrospray(NF-102,MECC Co.,Ogori,Japan)experimen-tal equipment consisted of a syringe pump,a stainless steel needle and a high voltage generator(Fig.1).An anionic or cationic poly-electrolyte aqueous solution was sprayed from a stainless steel needle(cathode)into an aqueous solution containing a polyelec-trolyte with an opposite charge(anode)in a foil-wrapped dish to form polyelectrolyte complex microcapsules.The polyelectrolyte aqueous solution in a dish was stirred continuously and gently (∼100rpm)by a magnetic stirrer bar during electrospraying.Typically,a sodium alginate(1.5wt%)aqueous solution(pH7.5) was sprayed into a chitosan aqueous(0.5wt%)solution(pH3.6) containing200mM acetic acid.The feed rate of the sodium alginate solution was set at0.20mL/h and the working voltage was22.5kV. The distance from the needle to the collector was5.0cm and the inner/outer diameters of a stainless steel needle were130/310m. Microcapsules were prepared under different conditions to control the diameters of microcapsules.After electrospraying,microcap-sules were separated from the chitosan solution by centrifugation at100×g for2min.An invertedfluorescence microscope(Olym-pus,IX71)was employed to observe microcapsules.Based on the microscope images,the diameters of more than100microcapsules Fig.1.Illustration of the experimental setup and photograph of a Taylor cone jet (inset).The Taylor cone was observed when a sodium alginate(1.5wt%)aque-ous solution was sprayed at0.20mL/h into a chitosan(0.5wt%)aqueous solution containing200mM acetic acid at a working voltage of22.5kV and a needle-to-collector distance of5cm.The inner/outer diameters of the stainless steel needle were130/310m.were measured by an image analysis software,WinROOF(Mitani Corp.,Fukui,Japan).PSS/PAH(PSS sprayed into PAH),PAH/PSS,chitosan/PSS and PDDA/PSS were also used for the preparation of microcapsules.The concentrations of PSS,PAH and PDDA were10wt%.The concentra-tion of chitosan solutions was9wt%.Only the chitosan solution contained50mM acetic acid.The pH of PSS,PAH,PDDA and chi-tosan solutions were4.8,1.6,2.6and6.5,respectively.The feed rate was0.20mL/h in each case,the voltage was24.0kV and the needle-to-collector distance was5.0cm.The inner/outer diameters of a stainless steel needle were330/630m.A Taylor cone,formed on the tip of a needle,was observed by a microscope(SKM-2000-PC,Saito Kougaku,Yokohama,Japan).2.3.Encapsulation of polymeric microspheres,protein anddextran and the dextran retention propertiesTo evaluate the encapsulation efficiency,fluorescent micro-spheres,albumin–FITC and TRITC–dextran were used as a core substrate.A sodium alginate(1.5wt%)aqueous solution contain-ing10L/mLfluorescent microspheres,10g/mL albumin–FITC or10g/mL TRITC–dextran was sprayed into0.5wt%chitosan aqueous solution containing200mM acetic acid under typical con-ditions.After spraying,a microcapsule suspension wasfiltered through a nylon-netfilter with a41m mesh(Millipore,NY). For the albumin–FITC encapsulation,thefiltrate was adjusted to pH6.0using sodium hydroxide.Non-encapsulatedfluorescence substrates in thefiltrate were quantified using afluorescence spectrometer(LS-50B,PerkinElmer).Fluorescence microspheres, albumin–FITC and TRITC–dextran were excited at468,495and 555nm,respectively.Fluorescence emissions were detected at 508,521and580nm,respectively.Thefluorescent microsphere-,albumin–FITC-and TRITC–dextran-encapsulated microcapsules were observed using a confocal laser scanning microscopy(CLSM) (FV1000-D,Olympus Co.,Tokyo,Japan).30Y.Fukui et al./Colloids and Surfaces A:Physicochem.Eng.Aspects370 (2010) 28–34To determine the dextran retention properties of the microcap-sules,TRITC–dextran-encapsulated microcapsules were collected by centrifugation at100×g for2min and dispersed in3M acetate buffer(pH5.2).Samples were periodically taken from the micro-capsule suspension and centrifuged at100×g.Thefluorescently labeled substrate in the supernatant solution was quantified using afluorescence spectrometer.2.4.Yeast-encapsulated microcapsulesThe yeast Saccharomyces cerevisiae Kyokai No.7was grown in a YPD medium(10g/L yeast extract,20g/L glucose,and20g/L peptone)at30◦C overnight.A sodium alginate(1.5wt%)aqueous solution containing yeast(OD∼0.35)was electrosprayed into a chi-tosan(0.5wt%)aqueous solution containing200mM acetic acid solution under typical conditions.The microcapsules were col-lected by centrifugation at100×g and then immersed in a fresh YPD medium,followed by microscope observation of the yeast growth in microcapsules at25◦C.3.Results and discussionWhen a polyelectrolyte solution is forced through a needle with an electric voltage,the surface tension of the solution becomes equal to the Coulomb repulsion.At this point,the solution at the tip of a needle forms a cone shape,called the“Taylor cone”[27]. If the electricfield intensity is larger than a balance point,small droplets are sprayed spontaneously from the tip of the Taylor cone into a counter electrode.As discussed widely in the literature,the formation of a Taylor cone is essential in electrospray and electro-spinning for the production of droplets andfibers.The formation of a Taylor cone generally requires certain operational conditions, such as particular feed rate,surface tension,conductivity,voltage, and so on[28–30].In the present study,a sodium alginate aque-ous solution was electrosprayed from a needle with a high voltage and the formation of a Taylor cone was also confirmed(inset of Fig.1),similar to previous reports[7,31,32].The charged droplets of the sodium alginate solution contacted the chitosan solution on the counter electrode and each immediately formed a polyelec-trolyte complex by an electrostatic interaction at the interface of the alginate solution/chitosan solution,resulting in a microcapsule. The alginate/chitosan microcapsules obtained by electrospray are shown in Fig.2a.The diameters of the microcapsules were from115 to145m,with a relatively narrow diameter distribution(Fig.2b), giving a coefficient of variation(CV)of only8.0%.These results indicate that the electrospray technique continuously produced aqueous droplets of a uniform size and that the droplets sprayed into the atmosphere reached the chitosan solution without expe-riencing droplet coalescence in the atmosphere during theirflight, probably due to the repulsion between charged droplets[33].We then investigated the effects of the operational conditions on the diameter of alginate/chitosan microcapsules.When varying one of the parameters,the other parameters were kept the same as those in Fig.2.Fig.3a shows the effect of feed rate on the diameter of the micro-capsules.The diameter of the microcapsules decreased from130 to80m with decreasing feed rate.The standard deviation also decreased with the diameter of microcapsules.Next,the effect of the working voltage on the diameter of microcapsules was stud-ied(Fig.3b).In the absence of a working voltage,the diameter of the microcapsules was around2.0mm,which agreed with previous reports[15,16].The electrostatic repulsions at the liquid level on the Taylor cone became stronger as the voltage increased,so that the droplets became smaller.As a result,the diameter of the result-ing microcapsules decreased with increasing voltage.As shownin Fig.2.(a)Microscope image and(b)size distribution of alginate/chitosan micro-capsules prepared by electrospray.Operating conditions:1.5wt%sodium alginate (0.2mL/h),0.5wt%chitosan,voltage22.5kV,needle-to-collector distance5cm,and inner/outer needle diameters130/310m.The scale bar represents100m.Fig.3c,we examined the effect of the needle-to-collector distance on the diameter of microcapsules.The diameter decreased with decreasing distance.This was probably due to the same reasons as given above for the effect of the working voltage:the shorter distance,the stronger the intensity of the electricfield.Fig.3d shows the effect of the inner diameter of the needle(130, 190,330and900m).The diameter of microcapsules increased linearly as the inner diameter of the needle increased from130to 330m.When a needle with an inner diameter of900m was used,the microcapsules were polydisperse(CV=39.3%).This is because the Taylor cone became unstable when large needle diam-eters were used.Interestingly,the diameters of the microcapsules were nearly the same as or smaller than the inner diameters of needles,even though there was negligible evaporation of water.In other meth-ods(e.g.,membrane emulsification,coacervation and microfluidic techniques),it was not possible to produce microcapsules smaller than the nozzle or pore diameter.This feature of the electrospray technique suggests the production of microcapsules and micro-spheres smaller than the limitations of microfabrication techniques is possible and this would be a great advantage in controlling the diameter of microcapsules and microspheres.The effect of the sodium alginate concentration(1.0,1.5,2.0wt%) was studied(Fig.3e).The pH of each sodium alginate solution was around7.5.At a sodium alginate concentration of1.0wt%, the mean diameter of microcapsules was95m with a CV of18%,Y.Fukui et al./Colloids and Surfaces A:Physicochem.Eng.Aspects370 (2010) 28–3431Fig.3.Effects of electrospray operating conditions on the diameter of microcapsules.(a)Feed rate,(b)working voltage,(c)needle-to-collector distance,(d)inner diameter of needle,(e)concentration of sodium alginate,(f)concentration of chitosan.Typical operating conditions were1.5wt%sodium alginate(0.2mL/h),0.5wt%chitosan,working voltage22.5kV and needle-to-collector distance5cm.Needle inner/outer diameters were130/310m.while the CV at1.5and2.0wt%were only9.9and9.3%,respec-tively.At0.5wt%,no spherical microcapsules were observed.When the concentration of sodium alginate is low,an alginate/chitosan complex would be formed but such a complex might be soluble in the chitosan solution because of the excess amount of polyca-tions present.The concentration of the chitosan solution,which received the droplets of alginate solution,also affected the diam-eters of microcapsules(Fig.3f).When the chitosan concentration was set at1.0,1.5and2.0wt%(pH3.9,4.1and4.3),the diame-ters of microcapsules were relatively small and there were many non-spherical microcapsules(inset of Fig.3f).The spherical micro-capsules were formed at0.5wt%chitosan solution(pH3.6).It can be presumed that the shape of microcapsules depends on the forma-tion rate and mechanical strength of the polyelectrolyte complex [34,35].There is room for further investigation on the mechanism of the microcapsule formation considering the formation rate and the mechanical strength of the polyelectrolyte complex.Not only the combination of alginate/chitosan but various other combinations of polyelectrolytes were also employed to pre-pare polyelectrolyte microcapsules.The combination of PSS/PAH (PSS sprayed into PAH),PAH/PSS,chitosan/PSS and PDDA/PSS were investigated(Fig.4).Although the combination of PSS/PAH (Fig.4d)also produced observable microcapsules,they had insuf-ficient mechanical strength for manipulation by pipetting.While a polyanion solution was sprayed into a polycation solution in the above investigations,we also sprayed a polycationic aque-32Y.Fukui et al./Colloids and Surfaces A:Physicochem.Eng.Aspects370 (2010) 28–34Fig.4.Microscope images of microcapsules produced from (a)PAH/PSS,(b)PDDA/PSS,(c)chitosan/PSS,(d)PSS/PAH.The concentrations of PAH,PSS and PDDA solutions were 10wt%and the concentration of chitosan solution was 9wt%,the feed rate was 0.20mL/h,the working voltage was 24.0kV and the needle-to-collector distance was 5.0cm.The inner/outer diameters of the needle were 330/630m.The scale bars represent 100m.ous solution into a polyanionic aqueous solution.As shown in Fig.4a,PAH/PSS microcapsules were successfully prepared.The mean diameter of the resultant microcapsules was 210m and the CV was 22%.Chitosan/PSS microcapsules (Fig.4c)were also prepared but were polydispersed.One of the reasons for the poly-dispersed chitosan/PSS microcapsules may have been that the lack of a balance of forces leading to an unstable Taylor cone,caused by high viscosity of the chitosan solution.The combination of PDDA/PSS (Fig.4b)produced microcapsules but their mechani-cal strength was as low as that of the PSS/PAH microcapsules (Fig.4d).Finally,we investigated the encapsulation of protein,dextran and cells in the polyelectrolyte microcapsules by electrospray.When protein,dextran and cells are encapsulated,the encapsula-tion efficiency and the residual activity are of considerable practical importance.In this electrospray technique,no organic solvent,heat or vacuum for drying are required.Therefore,it is expected that this method would be suitable for encapsulation of physi-ologically active substances and cells.In this study,fluorescent microspheres,albumin–FITC,TRITC–dextran and yeast cells were used as core substrates and a sodium alginate solution containing each core substrate was electrosprayed into a chitosan solution.Taking into account the capsule morphology,the productivity and stability of Taylor cone,the following production parameters were chosen to prepare alginate/chitosan microcapsules encapsulating the substrates;the concentrations of sodium alginate and chi-tosan were 1.5,0.5wt%,respectively,the feed rate of the sodium alginate solution was set at 0.20mL/h,the working voltage was 22.5kV,the distance from the needle to the collector was 5.0cm and the inner/outer diameters of a stainless steel needle were 130/310m.To approximate the encapsulation efficiency of yeast cells,we measured that of fluorescent microspheres because the size of fluorescent microspheres and yeast cells are nearly equal (a couple of micrometers).The CLSM images of the microcap-sules revealed that these fluorescent substances (the fluorescent microspheres and albumin–FITC)were uniformly spread over the whole microcapsules composed of alginate/chitosan,meaning that these substances were successfully encapsulated in the monodis-persed microcapsules (Fig.5).The encapsulation efficiency of microspheres and albumin was >99%.It should be noted that the diameters of the microcapsules and the size distribution were not affected by the presence of core substrates.These results demon-strate that the electrospray technique can encapsulate micro-and nano-sized materials in polyelectrolyte microcapsules with a high encapsulation efficiency.The retention of TRITC–dextran with different molecular weights in microcapsules was investigated.The retention ratios of all types of TRITC–dextran (Mw =4400,65,000–76,000and 155,000)in sodium alginate/chitosan microcapsules were >99%at 24h after dispersing microcapsules in an acetate buffer.Pre-vious studies reported that the ultrathin films of polyelectrolytes prepared by the layer-by-layer method displayed similarly high rejection properties to nanofiltration membranes [36–38].The high retention properties of the microcapsules in the present study were therefore reasonable.Fig.6shows phase-contrast microscope images of microcap-sules encapsulating yeast in a YPD medium at different periods after microcapsule preparation.As is evident from these images,yeast cells were also successfully encapsulated in the alginate/chitosan microcapsules and they grew inside the microcapsules over time.The image at 0h and the results from fluorescent microspheres allow us to speculate that most of cells sprayed were encapsulated within the microcapsules.The growth of yeast in a microcap-sule indicates sufficient penetration of low-molecular materials (glucose,peptone,etc.)from the culture medium through the microcapsule shell consisting of the alginate/chitosan complex.Even after 48h,microcapsules containing yeast cells did not tear,which indicates considerable mechanical strength of the algi-nate/chitosan microcapsules.These results demonstrate that the electrospray technique achieves the encapsulation of a physiolog-ically active substrate into polyelectrolyte microcapsules without any critical damage to the substrate.In summary,we prepared microcapsules based on cationic and anionic polyelectrolytes by the electrospray technique.The electrospray technique produced monodispersed alginate/chitosan microcapsules,whose size was controlled by varying the operatingY.Fukui et al./Colloids and Surfaces A:Physicochem.Eng.Aspects 370 (2010) 28–3433Fig.5.CLSM microscope images of alginate/chitosan microcapsules containing (a)and (b)fluorescent microspheres,(c)albumin–FITC,(d)TRITC–dextran (Mw =155,000).Operating conditions were the same as those in Fig.2.The scale bars represent 100m.conditions.This technique can utilize various types of natural and synthetic polyelectrolytes,and also encapsulate various substrates (biomacromolecules,microspheres and living cells)in the micro-capsules with high encapsulation efficiency and without critical damage to the substrates.Due to the simplicity of the electro-spray setup and the attractive properties discussed above,the electrospray technique is expected to be a practical method for the industrial production ofmicrocapsules.Fig.6.Phase-contrast images of yeast-encapsulated microcapsules in YPD media at different periods after the preparation of microcapsules.(a)0h,(b)8h,(c)16h,(d)24h.Operating conditions were the same as those in Fig.2.The scale bars represent 100m.34Y.Fukui et al./Colloids and Surfaces A:Physicochem.Eng.Aspects370 (2010) 28–34AcknowledgmentsWe thank Professor M.Kotaki at Kyoto Institute of Technology for the technical support.This work was supportedfinancially by Special Coordination Funds for Promoting Science and Technol-ogy,Creation of Innovation Centers for Advanced Interdisciplinary Research Areas(Innovative Bioproduction Kobe),MEXT,Japan.This work was partially supported by the Kao Foundation for Arts and Sciences.The present work was also partially supported by Grants for Research and Technology Development on Waste Management (K2119)from the Ministry of Environment,Japan.References[1]N.J.Zuidam,V.Nedovic,Encapsulation Technologies for Active Food Ingredientsand Food Processing,first ed.,Springer,2009.[2]M.Rosen,Delivery System Handbook for Personal Care and Cosmetic Products:Technology,Applications and Formulations,William Andrew,2006.[3]F.Lim,A.M.Sun,Microencapsulated islets as bioartificial endocrine pancreas,Science210(1980)908–910.[4]R.M.Shah,A.P.D’mello,Strategies to maximize the encapsulation efficiencyof phenylalanine ammonia lyase in microcapsules,Int.J.Pharm.356(2008) 61–68.[5]N.Gaponik,I.L.Radtchenko,M.R.Gerstenberger,Y.A.Fedutik,G.B.Sukho-rukov,A.L.Rogach,Labeling of biocompatible polymer microcapsules with near-infrared emitting nanocrystals,Nano Lett.3(2003)369–372.[6]L.Y.Chu,T.Yamaguchi,S.Nakao,A molecular-recognition microcapsule forenvironmental stimuli-responsive controlled release,Adv.Mater.14(2002) 386–389.[7]I.G.Loscertales,A.Barrero,I.Guerrero,R.Cortijo,M.Marquez,A.M.Ganan-Calvo,Micro/nano encapsulation via electrified coaxial liquid jets,Science295 (2002)1695–1698.[8]H.Zhang, E.Tumarkin,R.Peerani,Z.Nie,R.M.A.Sullan,G.C.Walker, E.Kumacheva,Microfluidic production of biopolymer microcapsules with con-trolled morphology,J.Am.Chem.Soc.128(2006)12205–12210.[9]Z.Nie,S.Xu,M.Seo,P.C.Lewis,E.Kumacheva,Polymer particles with variousshapes and morphologies produced in continuous microfluidic reactors,J.Am.Chem.Soc.127(2005)8058–8063.[10]B.Bugarski,Q.Li,M.F.A.Goosen,D.Poncelet,R.J.Neufeld,G.Vunjak,Electro-static droplet generation:mechanism of polymer droplet formation,AIChE J.40(1994)1026–1031.[11]V.A.Nedovic,B.Obradovic,I.Leskosek-Cukalovic,O.Trifunovic,R.Pesic,B.Bugarski,Electrostatic generation of alginate microbeads loaded with brewing yeast,Process Biochem.37(2001)17–22.[12]V.Manojlovic,J.Djonlagic,B.Obradovic,V.Nedovic,B.Bugarski,Investiga-tions of cell immobilization in alginate:rheological and electrostatic extrusion studies,J.Chem.Technol.Biotechnol.81(2006)505–510.[13]H.R.Brandenberger,F.Widmer,Immobilization of highly concentrated cell sus-pensions using the laminar jet breakup technique,Biotechnol.Progr.15(1999) 366–372.[14]J.W.Xie,C.H.Wang,Electrospray in the dripping mode for cell microencapsu-lation,J.Colloid Interface Sci.312(2007)247–255.[15]B.Philipp,H.Dautzenberg,K.J.Linow,J.Kotz,W.Dawydoff,Polyelectrolytecomplexes—recent developments and open problems,Prog.Polym.Sci.14 (1989)91–172.[16]R.Pommersheim,J.Schrezenmeir,W.Vogt,Immobilization of enzymes bymultilayer microcapsules,Macromol.Chem.Phys.195(1994)1557–1567.[17]G.Decher,Fuzzy nanoassemblies:toward layered polymeric multicomposites,Science277(1997)1232–1237.[18]K.C.Wood,J.Q.Boedicker,D.M.Lynn,P.T.Hammond,Tunable drug releasefrom hydrolytically degradable layer-by-layer thinfilms,Langmuir21(2005) 1603–1609.[19]H.Huang,E.Pierstorff,E.Osawa,D.Ho,Protein-mediated assembly of nanodi-amond hydrogels into a biocompatible and biofunctional multilayer nanofilm, ACS Nano2(2008)203–212.[20]D.Volodkin,Y.Arntz,P.Schaaf,H.Moehwald,J.C.Voegel,V.Ball,Compos-ite multilayered biocompatible polyelectrolytefilms with intact liposomes: stability and temperature triggered dye release,Soft Matter4(2008)122–130.[21]C.T.Yang,Y.Wang,S.Yu,Y.C.I.Chang,Controlled molecular organization ofsurface macromolecular assemblies based on stimuli-responsive polypeptide brushes,Biomacromolecules10(2008)58–65.[22]F.Caruso,R.A.Caruso,H.Mohwald,Nanoengineering of inorganic andhybrid hollow spheres by colloidal templating,Science282(1998)1111–1114.[23]F.Caruso,H.Lichtenfeld,M.Giersig,H.Mohwald,electrostatic self-assembly ofsilica nanoparticle polyelectrolyte multilayers on polystyrene latex particles,J.Am.Chem.Soc.120(1998)8523–8524.[24]S.Ye,C.Wang,X.Liu,Z.Tong,Multilayer nanocapsules of polysaccharidechitosan and alginate through layer-by-layer assembly directly on PS nanopar-ticles for release,J.Biomater.Sci.,Polym.Ed.16(2005)909–923.[25]K.Y.Lee,W.H.Park,W.S.Ha,Polyelectrolyte complexes of sodium alginate withchitosan or its derivatives for microcapsules,J.Appl.Polym.Sci.63(1997) 425–432.[26]O.Gaserod,O.Smidsrod,G.Skjak-Braek,Microcapsules of alginate–chitosan–I.A quantitative study of the interaction between alginate and chitosan, Biomaterials19(1998)1815–1825.[27]G.Taylor,Disintegration of water drops in an electricfield,Proc.R.Soc.Lond.A280(1964)383–397.[28]J.F.Delamora,I.G.Loscertales,The current emitted by highly conducting Taylorcones,J.Fluid Mech.260(1994)155–184.[29]R.P.A.Hartman,D.J.Brunner,D.M.A.Camelot,J.C.M.Marijnissen,B.Scarlett,Jetbreak-up in electrohydrodynamic atomization in the cone-jet mode,J.Aerosol Sci.31(2000)65–95.[30]A.M.GananCalvo,J.Davila,A.Barrero,Current and droplet size in the electro-spraying of liquids.Scaling laws,J.Aerosol Sci.28(1997)249–275.[31]R.Bocanegra,A.G.Gaonkar,A.Barrero,I.G.Loscertales,D.Pechack,M.Marquez,Production of cocoa butter microcapsules using an electrospray process,J.Food Sci.70(2005)492–497.[32]K.H.Roh, D.C.Martin,hann,Biphasic Janus particles with nanoscaleanisotropy,Nat.Mater.4(2005)759–763.[33]A.Jaworek, A.T.Sobczyk,Electrospraying route to nanotechnology:anoverview,J.Electrostat.66(2008)197–219.[34]V.D.Gordon,X.Chen,J.W.Hutchinson,A.R.Bausch,M.Marquez,D.A.Weitz,Self-assembled polymer membrane capsules inflated by osmotic pressure,J.Am.Chem.Soc.126(2004)14117–14122.[35]O.I.Vinogradova,O.V.Lebedeva,B.S.Kim,Mechanical behavior and character-ization of microcapsules,Annu.Rev.Mater.Res.36(2006)143–178.[36]W.Q.Jin,A.Toutianoush,B.Tieke,Use of polyelectrolyte layer-by-layer assem-blies as nanofiltration and reverse osmosis membranes,Langumir19(2003) 2550–2553.[37]S.U.Hong,ler,M.L.Bruening,Removal of dyes,sugars,and aminoacids from NaCl solutions using multilayer polyelectrolyte nanofiltration mem-branes,Ind.Eng.Chem.Res.45(2006)6284–6288.[38]S.U.Hong,R.Malaisamy,M.L.Bruening,Separation offluoride from othermonovalent anions using multilayer polyelectrolyte nanofiltration mem-branes,Langumir23(2007)1716–1722.。

Colloids and Surfaces B:Biointerfaces17(2000)145–152Effect of binders on polymorphic transformation kinetics of carbamazepine in aqueous solutionMakoto Otsuka*,Tomoko Ohfusa,Yoshihisa Matsuda Department of Pharmaceutical Technology,Kobe Pharmaceutical Uni6ersity,Motoyama-Kitamachi4-19-1,Higashi-Nada,Kobe658,JapanReceived7June1999;accepted9July1999AbstractThe effects of binders on the polymorphic transformation kinetics of carbamazepine(CBZ)were investigated by thermal analysis and X-ray diffraction analysis.The binders used were hydroxypropylcellulose(HPC)(HPC-SL, molecular weight30000–50000;HPC-M,molecular weight50000–70000;HPC-L,molecular weight110000–150000).CBZ anhydrate form I and various concentrations of binder solutions were mixed at1000rpm and25°C. The amount of dihydrate transformed was evaluated based on the latent heat due to dehydration on DSC curves. Since thefirst-order plots for transformation process of CBZ showed a straight line,the transformation rate constant, k and induction period,IP were estimated based onfirst-order kinetics by the least-squares method.The k of CBZ decreased with increase of HPC-L concentration,but the IP increased.In contrast,the k of phase transformation on addition of crystal seeds was almost the same as that without seeds,but the IP significantly decreased on seed addition.The result suggested that IP was a nucleus formation process,but the seed addition did not affect the crystal growth process.The molecular weight effect of HPC on the transformation suggested that the k of HPC-SL was the largest,with the rank order being HPC-SL\HPC-M\HPC-L.The order for IP was HPC-L\HPC-SL\HPC-M. The relation between IP and kinematic viscosity had a straight line,but the k decreased with increase of kinematic viscosity.The increase of IP on addition of HPC might be induced by inhibition of the formation of nuclei by the steric intermolecular effect of HPC and decrease of D v.Therefore,HPC strongly inhibited nucleus formation in the crystallization of CBZ.©2000Elsevier Science B.V.All rights reserved.Keywords:Polymorphic transformation;Carbamazepine;Hydration;Nucleation;Crystal growth;Hydroxypropylcellulosewww.elsevier.nl/locate/colsurfb1.IntroductionHigh quality granular preparations offer a number of potential advantages to the pharma-ceutical industry in the production of beads or granules as bothfinished and intermediate prod-ucts.Therefore,various kinds of granulation tech-niques and equipment,such as extruders,fluidized beds and high speed mixers,have been developed to obtain high quality granular materials[1].Con-trolling these properties is an important factor in the production of high quality pharmaceuticals.*Corresponding author.Tel.:+81-78-441-7531;fax:+81-78-441-7532.E-mail address:m-otsuka@kobepharma-u.ac.jp(M.Otsuka)0927-7765/00/$-see front matter©2000Elsevier Science B.V.All rights reserved. PII:S0927-7765(99)00111-3M.Otsuka et al./Colloids and Surfaces B:Biointerfaces17(2000)145–152 146However,the pharmaceutical properties of gran-ules depend on various physical and chemical factors during manufacturing processes,such as instruments,formulations and manufacturing conditions.There is a few studies concerning to crystalline transformations during manufacturing process.The polymorphic form of an insoluble drug influences the bioavailability of preparations by affecting the dissolution rate[2].The physico-chemical stability related polymorphism affects the pharmaceutical properties,such as disintegra-tion time and mechanical strength of the prepara-tion[3].Carbamazepine(CBZ)is widely used as a potent anticonvulsant,and there have been re-ports concerning its polymorphic form[4–6]. CBZ polymorphic transformation at high humid-ity[7]and in aqueous suspension[8]was investi-gated and it was concluded that all forms transformed into the dihydrate.However,in a previous study[9],CBZ anhydrate form I was not transformed into dihydrate during CBZ granular formulation.This suggested that excipients,such as a binder,diluent and disintegrator,interacted with CBZ,and inhibited CBZ polymorphic trans-formation during granulation.In this study,there-fore,to clarify the interaction between excipients and CBZ,we investigated the effect of HPC as a binder on the polymorphic transformation of CBZ anhydrate.2.Materials and methods2.1.MaterialsCBZ bulk powder of Japanese Pharmacopoeia (JP)XIII grade(lot No.CEE-9-5)was obtained from Katsura Chem.Co.,Tokyo,Japan.The bulk powder was identified as being of polymor-phic form I[7]by X-ray diffraction analysis and DSC measurement.Hydroxypropylcellulose (HPC)(HPC-SL,molecular weight30000–50000;HPC-M,molecular weight50000–70000; HPC-L,molecular weight110000–150000;Ni-hon Soda Co.Japan)was used as a binder.All other chemicals were of analytical grade.2.2.Preparation of polymorphsThe CBZ bulk powder was identified as being of form I(anhydrate,monoclinic CBZ)[7].The dihydrate form(form IV)[7]was obtained by recrystallization as follows:the CBZ bulk powder was dissolved in50%ethanol solution in a water bath at70°C,andfiltered.After cooling the satu-rated CBZ solution to room temperature,the crystalline samples werefiltered and dried in a desiccator containing silica gel at room tempera-ture under vacuo for3h.All of the CBZ samples were passed through a no.200mesh(75m m) screen.2.3.Viscosity measurementThe kinematic viscosity was determined using a capillary viscometer at25°C(JP XIII).2.4.X-ray powder diffraction analysisDiffractograms were taken at room tempera-ture with an X-ray diffractometer(XD-3A,Shi-madzu Co.,Kyoto,Japan).The operating conditions were as follows:Target,Cu;filter,Ni; voltage20kV,current,20mA;receiving slit,0.1 mm;time constant,1s;counting range,1kcps; scanning speed4°2q min−1.2.5.Thermal analysisDifferential scanning calorimetry(DSC)was performed with a type3100instrument(Mac Sci-ence Co.,Tokyo).The operating conditions in the open-pan system were as follows:sample weight, 5mg;heating rate,10°C min−1;N2gasflow rate, 30ml min−1.2.6.Polymorphic transformation processA total of30g of CBZ bulk powder was mixed with200ml of0,0.001,0.005,0.05w/v%binder solution in a1000-ml round bottomedflask at a mixing speed of1000rpm at25°C.To evaluate the amount of dihydrate transformed5ml of suspension was sampled during mixing at5,10, 20,30,4560and85min,and afterfiltering theM .Otsuka et al ./Colloids and Surfaces B :Biointerfaces 17(2000)145–152147Fig.1.Powder X-ray diffraction profiles of CBZ forms.w /w%crystal content)in a mortar.The DSC curves of the standard samples were measured in triplicate.The standard deviation of the data was within 5%.The plots gave a good linear correla-tion and the linear regression equations were as follows:Y =308.4X +5.8(R =0.998)(1)where Y (J g −1)is a latent heat due to dehydra-tion of dihydrate CBZ.X is the dihydrate concen-tration (w /w%).R is the correlation coefficient.3.Results3.1.Effect of HPC -L concentration in binder solution on CBZ phase transformationFig.1shows the X-ray diffraction profiles of CBZ anhydrate,form I and dihydrate,form IV.The X-ray diffraction patterns of form I and dihydrate were significantly different and identical to those reported [6,7].Fig.2shows the effect of HPC-L concentration on the endothermic peak due to dehydration on DSC curves of CBZ dihy-drate.The endothermic peak of the samples in 0and 0.001%binder solution increased with mixing time,but not so in 0.05%solution.The X-ray diffraction profiles were consistent with this resultwet powders were stored at 30°C,47%RH for 24h to measure DSC.2.7.Measurement of the dihydrate contentThe dihydrate (form IV)content of the samples was measured by DSC based on the endothermic peak at around 70°C due to dehydration.Briefly,known quantities of standard mixtures were ob-tained by physically mixing anhydrate and dihy-drate at various ratios (0,25,50,75and 100Fig.2.Change of the DSC curves of CBZ forms during mixing (1000rpm)in binder solution at 25°C.M.Otsuka et al./Colloids and Surfaces B:Biointerfaces17(2000)145–152 148Fig.3.Effect of HPC-L concentration on hydration of CBZ form I. ,without additive; ,0.001%HPC-L; ,0.005% HPC-L; ,0.05%HPC-L.Fig.4.Effects of HPC-L concentration and the addition of seed crystals on thefirst-order plots for the hydration of CBZ form I. ,without additive; ,0.001%HPC-L; ,0.005% HPC-L; ,0.05%HPC-L; ,0.005%HPC-L with0.5% seeds.3.2.Effect of the molecular weight of the binder on CBZ phase transformationFig.5shows the effect of the molecular weight of HPC on CBZ phase transformation in aqueous suspension at25°C.The k and IP are summarized in Table1.The k of HPC-SL was the largest,with the rank order being HPC-SL\HPC-M\HPC-L.The(the data not shown).Thus,the CBZ anhydrate appeared to be transformed immediately to dihy-drate in aqueous suspension,but not in high binder solution.The amount of dihydrate transformed was evaluated based on the latent heat due to dehydration on DSC curves.Fig.3shows the effect of HPC-L concentration on the trans-formation of CBZ dihydrate.This suggested HPC-L inhibited the transformation of di-hydrate,and the effect depended on the concen-tration.Fig.4shows the effect of HPC-L on the first-order plots for polymorphic transformation of form I.Since the plot shows a straight line, the transformation rate constant,k and induction period,IP were estimated based onfirst-order kinetics by the least-squares method.The results are summarized in Table1.The k of CBZ de-creased with the increase of HPC-L concentra-tion,but the IP increased.In contrast,the k of phase transformation on the addition of crystal seeds was almost the same as that without seeds, but the IP significantly decreased on seed addi-tion.Table1Kinetic parameters for the transformation of CBZ aIP(min)k(min−1)Sample V(mm−2s−1)1.06×10−1 1.40 1.0000%binder0.001%HPC-L8.305.70×10−2 1.0060.005%HPC-L29.11.10×10−2 1.0261.04960.00.050%HPC-L02.63×10−226.70.005%HPC- 1.008SL0.005%HPC-14.76.10×10−3 1.018M1.0267.1431.41×10−20.005%HPC-L+seedsa k,transformation rate constant;IP,induction period;V, kinematic viscosity at25°C.M.Otsuka et al./Colloids and Surfaces B:Biointerfaces17(2000)145–152149 Fig.5.Effects of the molecular weight of HPC on thefirst-or-der plots for the hydration of CBZ form I. ,withoutadditive; ,0.005%HPC-SL; ,0.005%HPC-M; ,0.005%HPC-L.Fig.6.Effects of kinematic viscosity on the IP for hydration ofCBZ form I. ,without additive; ,HPC-L; ,HPC-M; ,HPC-SL.rank order for IP was HPC-L\HPC-SL\HPC-M.Since HPC-L has the highest molecular weight,the rank is HPC-L\HPC-M\HPC-SL, thus the k and IP were affected by the molecular weight of HPC.3.3.Effect of kinematic6iscosity on CBZ phase transformationFigs.6and7shows the effect of kinematic viscosity on the IP and k of CBZ transformation. The relation between IP and kinematic viscosity had a straight line,but that of k was a nonlinear and the k decreased with increase of kinematic viscosity.This suggested that the viscosity of the solution affected the crystal transformation kinet-ics of CBZ.4.Discussion4.1.Crystalline formation kineticsIn general,crystalline growth kinetics[11]con-sisted of nucleation and crystal growth and was controlled by various physical and chemical fac-tors.Eq.(4)shows nucleation velocity(J)based on Gibbs free energy change(D G)at constant temperature and pressure.The D G for formation of nuclei in radius r is expressed in Eq.(2),and dependent on D v and surface tension k.D G*is the formation of nuclei of clinical radius(r*) Fig.7.Effects of kinematic viscosity on the k for hydration of CBZ form I. ,without additive; ,HPC-L; ,HPC-M; , HPC-SL.M .Otsuka et al ./Colloids and Surfaces B :Biointerfaces 17(2000)145–152150Fig.8.Simulation curve of D G and J for the formation of nuclei upon crystallization based on Eqs.(3)and (4).The solid line and dotted lines represent D G and J ,respectively.The solubility is 10.ation,J ,increases with increase of D v ,the degree of supersaturation of solution,indicating that D v affects the D G and J of the crystallization process.Fig.9shows the effect of D v on the R max /J ratio of crystallization.The profile shows a maxi-mum peak at 1.3,and a decrease at values greater than D v =1.3(solubility =10).This suggests that the nucleation proceeds at higher D v ,but the crystal growth occurs at an D v of around 1.3after nucleation.Thus both nucleation and crystal growth proceed in more supersaturated solutions,but crystal growth occurs after nucleation at a lower degree of supersaturation.Young and Suryanarayanan [8]reported that CBZ anhydrate (form I)transformed into dihy-drate in aqueous solution following a first-order kinetics.In the present study,the crystalline trans-formation of CBZ anhydrate in aqueous solution also apparently followed first-order kinetics (Fig.4).However,the solid-to-solid transformation of CBZ during granulation is not simple crystalliza-tion,because it includes a starting material disso-lution process.In general,dissolution follows the Noiyes–Nernst Eq.(6).Kaneniwa et al.,[5]re-ported dissolution kinetics of CBZ form I based on the diffusion controlled dissolution model as shown in Eq.(6).−d C d t =DS wV l (C s−C )(6)D ,diffusivity of the drug;S w ,specific surface area;V ,volume of solution;t ,thickness of diffu-sion layer;C s ,solubility,C ,drug concentration at time t .obtained in Eq.(3).The J is affected through D G *by D v and k .D G (r )=4y r 33 D v +4y r 2k(2)D G*=4·4yk 3 23D v 2(3)J =4y r *2p q 1exp−D G*kT(4)where k ,surface energy; ,molecular volume;D v ,the degree of supersaturation of solution;s ,Boltzmann’s constant;m ,mass;r *,critical radius of nuclei;T ,absolute temperature;p ,concentra-tion of solution;q 1,density of critical nuclei at 0K.The crystal growth kinetics [11,12]is expressed as Wilson–Frenkel’s Eq.(5).Crystal growth rate,R max ,is also dependent on D v but independent of surface tension.R max = c 6aC e exp−D G dsl 8s T log(1+l )=D v(5)R max ,crystal growth rate; c ,molecular volume of crystal,a ,distance of crystal lattice;w ,vibration number of molecule;C e ,solubility of solution;D G ds ,Gibbs free energy of desolvation;|,degree of supersaturation;m ,mass.Fig.8shows the simulated results for nucle-ation based on Eqs.(3)and (4)at a constant temperature and pressure.The velocity of nucle-Fig.9.Simulation curve of D G /J for the formation of nuclei on crystallization based on Eqs.(3)and (4).The solubility is 10.M .Otsuka et al ./Colloids and Surfaces B :Biointerfaces 17(2000)145–152151Since there are several rate determination steps for crystalline transformation of CBZ,the crys-talline transformation in a suspension or during the kneading of wet mass for granulation consists of dissolution,nucleation and crystal growth as shown.Anhydrate Dissolutionk dSupersaturated solution Nucleationk nSS* Crystal growthk cDihydrate k d is the dissolution coefficient constant,k n is the nucleation rate constant and k c is the crystal growth rate constant.The SS*is activated super-saturated solution containing enough nuclei to start the crystal growth.Thus,the crystal trans-formation followed consecutive reaction kinetics,and included an induction period.4.2.Kinetic mechanism of inhibition of CBZ phase transformationIt is well known that polymer inhibits crystal growth in solution.Watanabe et al.[10]reported the inhibitory effect of sodium caseinate on the recrystallization of pheytoin from supersaturated solution.They concluded that the crystalline growth of phenytoin was inhibited by depression of the nuclei formation rate.On the other hand,the crystalline transformation of CBZ anhydrate apparently followed first-order kinetics,and was consistent with previous results.However,CBZ anhydrate in a formulation containing 0.05%HPC did not transform into dihydrate during granulation.Since the change of IP relative to viscosity was a linear (Fig.6),but that of k was non-linear (Fig.7),the mechanism of IP prolongation is not the same as that of depression of k .HPC might interact with CBZ,and inhibit crystallization of CBZ.On the other hand,the increase of viscosity on addition of HPC also might affect drug diffusion,D ,in dissolution (Eq.(6))and the activation energy of drug diffusion (D G ds )in crystal growth (Eq.(4)).This suggests that on increase of viscosity would induce a decrease of D v in the solution,and affect both the nucleation and crystal growth process.The results the effects of HPC con-centration (Fig.4)and molecular weight (Fig.5)effects are almost supported the theoretical hy-pothesis.The role of fine dihydrate crystals was investi-gated (see Fig.4).The k of 0.5%with seeds was almost the same as that without seeds,but the IP was significantly decreased by the seeds addition.The result suggested that IP was the nucleusformation process,but the seed addition is not affected by crystal growth.The increase of IP on addition of HPC (Fig.6)might be induced by inhibition of the formation of nuclei by the steric intermolecular effect of HPC and decrease of D v .Therefore,the addition of seed crystals did not affect the crystal growth rate.In other words,HPC strongly inhibited nucleus formation in the crystallization of CBZ.5.ConclusionsThe transformation of CBZ anhydrate was de-pressed dependent on the binder solution concen-tration.The transformation inhibition might be caused by the reduced of nucleus formation on HPC addition.The nature of the bulk powders and excipients,such as the polymorphic form,particle size and distribution,crystallinity etc.,reflect the history of the chemical and physical treatments of the raw materials.Since the phar-maceutical properties of the preparation are changed by the interaction between the formula-tions during the manufacturing process,the phar-maceutical properties could be controlled by regulating of the formula of the excipients.There-fore,to prepare better quality granules it is neces-sary to monitor and control the characteristics of the powder materials,such as bulk and excipient powders.AcknowledgementsThis work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education,Science and Culture,Japan.M.Otsuka et al./Colloids and Surfaces B:Biointerfaces17(2000)145–152 152References[1](a)M.Otsuka,J.Gao,Y.Matsuda,Effect of amount ofadded water during exrusion–spheronization process of pharmaceutical properties of granules,Drug Dev.Ind.Pharm.20(1994)2977–2992.(b)C.W.Woodruff,N.O.Nuessle,Effect of processing variable on particles ob-tained by extrusion–spheronization processing,J.Pharm.Sci.61(1972)787–790.(c)S.Watano,A.Yamamoto,K.Miyanami,Effect of operational variables on the proper-ties of granules prepared by moisture control method in tumblingfluidized bed granulation,Chem.Pharm.Bull.42(1994)133–137.(d)T.Shiraishi,S.Kondo,H.Yuasa, Y.Kanaya,Studies on the granulation process of gran-ules for tableting with high speed mixer.I.Physical properties of granules for tableting,Chem.Pharm.Bull.42(1994)932–936.[2](a)A.E.A.R.Ebian,R.M.A.Moustafa,E.B.Abul-Enin,Nitrofurantoin I.Effect of aging at different relative humidities and higher temperatures on the drug release and the physical properties of tablets,Egypt.J.Pharm.Sci.26(1985)287–300.(b)A.E.A.R.Ebian,H.T.Fikrat, R.M.A.Moustafa, E.B.Abul-Enin,Nitrofurantoin II.Correlation of in vivo bioavailability to in vitro dissolu-tion of nitrofurantoin tablets aged at different relative humidities and elevated temperatures,Egypt.J.Pharm.Sci.27(1986)347–358.(c)FDA Paper,Guideline:Manu-facturing and Controls for INDs and NDAs,Pharm.Tech.Jpn.1(1985)835.[3](a)J.K.Haleblian,Characterization of habits and crystal-liine modification of solids and their pharmaceutical ap-plications,J.Pharm.Sci.64(1975)1269–1288.(b)H.W.Gouda,M.A.Moustafa,H.I.Al-Shora,Effect of storage on nitrofurantoin solid dosage forms,Int.J.Pharm.18 (1984)213.[4](a)H.Po¨lmann,C.Gulde,R.Jahn,S.Pfeifer,Polymor-phie,Teilchengru¨e und Blutspiegelwerte von Carba, azepin,Pharmazie30(1975)709–711.(b)C.Lefebvre,A.M.Guyot-Hermann,M.Draguet-Brughmans,R.Bouche´,Polymorphic transformations of carbamzepine during grinding and compression,Drug Dev.Ind.Pharm.12(1986)11–13.[5]N.Kaneniwa,J.Ichikawa,T.Yamaguchi,K.Hayshi,N.Watari,M.Sumi,Dissolution behavior of carba-mazepine polymorhps,Yakugaku Zasshi107(1987)808–813.[6]P.Kahela,R.Aaltonen,E.Lewing,M.Anttila,E.Krist-offersson,Pharmacokinetics and dissolution of two crys-talline forms of carbamzepine,Int.J.Pharm.14(1983) 103–120.[7]N.Kaneniwa,T.Yamaguchi,N.Watari,M.Otsuka,Hygroscopicity of carbamazepine crystalline powders, Yakugaku Zasshi104(1984)184–190.[8]W.W.L.Young,R.Suryanarayanan,Kinetics of transfor-mation of anhydrous carbamazepine to carbamazepine dihydrate in aqueous suspensions,J.Pharm.Sci.80 (1991)496–500.[9]M.Otsuka,H.Hasegawa,Y.Matsuka,Effect of poly-morphic transformation during the extrusion-granulation process on the pharmaceutical properties of carba-mazepine granules,Chem.Pharm.Bull.45(5)(1997) 894–898.[10]A.Watanabe,S.Suzuki,M.Sugihara,Inhibitory effect ofsodium caseinate on the phenytoin recrystallization, Yakuzaigaku50(1990)179–186.[11]T.Kuroda,Crystal is Living,Saiensu-Sya,Tokyo,Japan,1984.[12]Hand Book of Crystalline Technology,in:M.Yamamoto(Ed.),Kyoritsu Press,Tokyo,Japan,1971..。

Measurement of the corrosion rate of magnesium alloys using Tafel extrapolationZhiming Shi,Ming Liu,Andrej Atrens *The University of Queensland,Division of Materials,Brisbane,Qld 4072,Australiaa r t i c l e i n f o Article history:Received 13July 2009Accepted 9October 2009Available online 6November 2009Keywords:A.Magnesium corrosionB.Weight lossB.Tafel extrapolation B.Hydrogen evolutiona b s t r a c tThe hypothesis that the corrosion of Mg alloys can be adequately estimated using Tafel extrapolation of the polarisation curve is termed herein the electrochemical measurement hypothesis for Mg.In principle,such a hypothesis can be disproved by a single valid counter example.The critical review of Mg corrosion by Song and Atrens in 2003indicated that,for Mg alloys,Tafel extrapolation had not estimated the cor-rosion rate reliably.This paper examines the recent literature to further examine the electrochemical measurement hypothesis for Mg.The literature shows that,for Mg alloys,corrosion rates evaluated by Tafel extrapolation from polarisation curves have not agreed with corrosion rates evaluated from weight loss and hydrogen evolution.Typical deviations have been $50–90%.These were much larger than the precision of the measurement methods and indicate a need for careful examination of the use of Tafel extrapolation for Mg.For research that nevertheless does intend to use Tafel extrapolation to elucidate corrosion of Mg associated with service,it is strongly recommended that these measurements be com-plemented by the use of at least two of the three other simple measurement methods:(i)weight loss rate,(ii)hydrogen evolution rate,and (iii)rate of Mg 2+leaving the metal surface.There is much better insight for little additional effort.Ó2009Elsevier Ltd.All rights reserved.1.IntroductionMagnesium (Mg)alloys are used in transport applications,such as in auto construction,because of their low density,adequate strength–weight ratio and excellent castability.However,an issue is their corrosion properties [1–5].As a consequence,there is much current research to understand Mg corrosion for such ser-vice applications.Some of this research relies on the measurement of the corrosion rate using Tafel extrapolation from polarisation curves.Such research relies on what is herein termed the electro-chemical measurement hypothesis for Mg,namely,that the corro-sion rate of Mg alloys can be adequately estimated using Tafel extrapolation of the polarisation curve.In principle,such a hypoth-esis can be disproved by a single valid counter example.The critical review of Mg corrosion by Song and Atrens [2]indicated that,for Mg alloys,Tafel extrapolation had not estimated the corrosion rate reliably.The scope of this paper is to examine the recent literature to further examine the electrochemical measure-ment hypothesis for Mg.A subsidiary aim is to facilitate research directed at Mg alloy development and at understanding corrosion of Mg in service applications to ensure such research is as effective as possible.2.Corrosion rate measurementThe simplest and most fundamental measurement of the corro-sion rate is the metal weight loss rate,D W (mg/cm 2/d).This can be converted to an average corrosion rate (mm/y)using [6–9]P W ¼3:65D W =qð1Þwhere q is the metal density (g/cm 3).For Mg alloys,q is 1.74g/cm 3,and Eq.(1)becomes:P W ¼2:10D W ð2ÞIn the overall corrosion reaction of pure Mg,one molecule of hydro-gen is evolved for each atom of corroded Mg.One mol (i.e.24.31g)of Mg metal corrodes for each mol (i.e.22.4L)of hydrogen gas pro-duced.Therefore,the hydrogen evolution rate,V H (ml/cm 2/d),is re-lated to the metallic weight loss rate,D W m (mg/cm 2/d),using [2,10–14]D W ¼1:085V H ð3ÞThe corresponding corrosion rate,P H ,is evaluated by substituting Eq.(3)into Eq.(2)to giveP H ¼2:279V Hð4ÞFor Mg corrosion,there is excellent agreement [2,10,13,15]between the corrosion rate measured by the weight loss rate and that eval-uated from the hydrogenevolution rate.Fig.1presents a cross plot0010-938X/$-see front matter Ó2009Elsevier Ltd.All rights reserved.doi:10.1016/j.corsci.2009.10.016*Corresponding author.Address:The University of Queensland,Division of Materials,St.Lucia,Brisbane,Qld 4072,Australia.Tel.:+61733653748;fax:+61733653888.E-mail address:andrejs.atrens@.au (A.Atrens).Corrosion Science 52(2010)579–588Contents lists available at ScienceDirectCorrosion Sciencej o ur na l h om e pa ge :w w w.e lse v ie r.c om /lo c at e /c or s c iof the independent measurements of the weight loss rate and the hydrogen evolution rate,for48h and96h immersion tests of castAZ91in1M NaCl solution[13].The line,in Fig.1,is a goodfit through the data.However,the line is not drawn as a line of best fit through the data.The line is actually the plot of the theoretical expectation of Eq.(3).This data shows that the corrosion rate eval-uated from weight loss agrees within an error of$±10%with the corrosion rate independently measured from hydrogen evolution, as also indicated by the review of Song and Atrens[2].In the Tafel extrapolation method for measuring the Mg corro-sion rate,the corrosion current density,i corr(mA/cm2)is estimated by Tafel extrapolation of the cathodic branch of the polarisation curve,and i corr is related to the average corrosion rate using [6–9,11,14]P i¼22:85i corrð5ÞReasons were given by Song and Atrens[2]why this electrochemi-cal technique might not give reliable values for Mg corrosion.Nev-ertheless the electrochemical technique of Tafel extrapolation is widely used for the evaluation of the corrosion of Mg alloys,at least partly,because it is a quick and easy technique.Therefore it is use-ful to review the literature on this technique for Mg alloys.It is useful to have quantitative measures of the quality of the corrosion rate evaluated by the Tafel extrapolation.One measure is to evaluate the relative deviation between the measured corro-sion rate and a standard corrosion rate.The corrosion rate evalu-ated from weight loss,P W,and the corrosion rate,P H,evaluated from the hydrogen evolution data,can each be used as a standard against which to compare the corrosion rate evaluated by Tafel extrapolation.Experimentally it has been shown that there is good agreement between measurements of P W and P H.The relative devi-ation for the corrosion rate determined from Tafel extrapolation was evaluated usinge i¼P iÀP WðÞ=P WðÞj jÂ100ð6Þore i¼P iÀP HðÞ=P HðÞj jÂ100ð7ÞAn alternative measure of quality is the ratio P W/P i.This ratio would be equal to unity if P i were a good estimate of P W.Mg corrosion has a strange phenomenon that anodic polarisation increases both the amount of Mg2+ions produced AND the amount of hydrogen evolved[1–3].This is summarised in the Section5.The most likely explanation is that part of the corrosion reaction is chemical rather than electrochemical.The consequence is that the ratio P W/P i would be expected to have a value between1and2,if P W is a good measure of the rate of the total corrosion reaction and if P i were a good measure of the rate of the electrochemical part of the total corrosion reaction.3.Corrosion rate measurements from Tafel extrapolation3.1.Pure Mg,AZ91and ZE41in1M NaClTable1[15]relates to the corrosion of pure Mg,AZ91and ZE41 in1M NaCl.All three alloys were as cast and all were high purity in that the concentration of the impurity was lower than the impurity tolerance limits[16].AZ91and ZE41are both two-phase alloys;in each case,the second phase was not continuous so there was no tendency for the second phase to provide a barrier effect [2,13,17].Samples were encapsulated in resin and only one surface was exposed to the solution.This approach facilitated the exami-nation of the corrosion morphology.The specimen surface was mechanically ground to1200grit on SiC paper,washed with dis-tilled water and dried.The hydrogen evolution was measured overa period of48h using duplicate specimens exposed horizontally to1.5L of1M NaCl.For ZE41the corrosion initiated as localized cor-rosion at some sites on the surface and subsequently expanded over the whole surface.The hydrogen evolution,after an initiation time of several hours,increased linearly with exposure time.The advance of the corrosion over the surface of AZ91was slower, although the corrosion also initiated as localized corrosion;there was corrosion on only part of the surface at the end of the48h per-iod.The hydrogen evolution,after an initiation time of several hours,increased with exposure time;the hydrogen evolution rate increased slightly with time.Nevertheless,the hydrogen evolution volume for AZ91was significantly lower than for ZE41.The corro-sion of the pure Mg was uniform over a macroscale in that there were no preferential sites for corrosion.At the end of the48h exposure,the corrosion was essentially uniform over the whole exposed surface area.The hydrogen evolution,after an initiation time of several hours,increased linearly with exposure time.The hydrogen evolution volume for pure Mg was significantly lower than for the two alloys,consistent with the general observation that the lowest corrosion rate is produced by pure Mg[1,2]and that the higher corrosion rates of the two alloys was in each case due to micro-galvanic acceleration of the corrosion by the second phase.The average hydrogen evolution rate over the exposure period is reported in Table1[15].At the end of the exposure per-iod,the corrosion products were removed by the standard cleaning solution,and the weight loss evaluated.The weight loss rate is also contained in Table1.There was good agreement between the corrosion rate evaluated from the hydrogen evolution rate and that evaluated from the weight loss rate,as expected from Fig.1and from the review of Song and Atrens[2].The polarisation curves were measured in a standard three-electrode glass cell with a standard scan rate of0.2mV/s.The reference electrode was a saturated calomel electrode(SCE).The specimen configuration was the same as that used in the immer-sion experiments,which measured the hydrogen evolution and the weight loss.Duplicate polarisation curves were essentially identical.In each case the cathodic branch provided an extensive linear Tafel region,the evaluated i corr value is included in Table1 as is the corresponding corrosion rate.The corrosion rate values derived from Tafel extrapolation were quite different to those derived from the hydrogen evolution rate and from the weight loss data.The lower derived corrosion rates for ZE41and AZ91might be attributed to the incubation of corrosion as shown by the lower hydrogen evolution rates at the beginning of the immersion tests.Fig. 1.Cross plot of the independent measurements of weight loss rate andhydrogen evolution rate,for48h and96h immersion tests of cast AZ91in1M NaClsolution.The line is a plot of the theoretical expectation;it is not simply a linethrough the experimental data points.580Z.Shi et al./Corrosion Science52(2010)579–588The corrosion rate for pure Mg from the Tafel extrapolation was about three times that measured by weight loss or hydrogen evolution.This might be associated with the transformation of an air formedfilm to the steady statefilm for Mg exposed to water [18].However,the hydrogen evolution data did not show any analogous feature,the hydrogen evolution was low and,as far as could be discerned,the hydrogen evolution was linear with time.The data of Table1relates to the corrosion rate,P W and P H,mea-sured over48h and P i measured soon after specimen immersion in the solution.For all three Mg alloys,there was a large relative deviation for the corrosion rate determined from Tafel extrapola-tion compared with the weight loss rate.The relative deviation for the corrosion rate determined from Tafel extrapolation was calculated using Eq.(6).This large relative deviation is in agree-ment with the observations of the review of Song and Atrens[2].3.2.Heat Treated Mg–10Gd–3Y–0.4Zr in5%NaClTable2relates to the study of Peng et al.[19];they studied the corrosion of Mg alloy Mg–10Gd–3Y–0.4Zr,in the as cast(F),solu-tion treated(T4)and aged(T6)conditions,in5%NaCl solution, by immersion tests and potentiodynamic polarisation curves.The corrosion rate was measured using three or four replicate speci-mens,for each microstructure condition,immersed for3days at 25±2°C in5wt.%NaCl aqueous solution,prepared with AR grade NaCl and distilled water.Each specimen was polished successively on320grit waterproof abrasive paper and three grit metallo-graphic paper,washed with distilled water,dried in warmflowing air and weighed to determine the original weight.During thefirst few hours of immersion,the solution pH increased from neutral to pH$11due to the precipitation of Mg(OH)2because of its low solubility.Thereafter the pH remained constant at$11.Because the time of the initial increase in pH was short(a few hours)com-pared with the test duration(3days)the corrosion rates measured were essentially the same as measured in a solution saturated with Mg(OH)2.After the immersion test,each specimen was washed with distilled water and dried.One sample was used for the corro-sion product analysis.The other samples were used to determine the corrosion rate by means of the metal weight loss.The corrosion products were removed by sample immersion in a solution of 200g LÀ1CrO3+10g LÀ1AgNO3at ambient temperature for 7min.Separate experiments demonstrated that this treatment re-moves all the corrosion products without removing any Mg metal. Each specimen was then washed with distilled water,dried in the warmflowing air and weighed to determine the weight after cor-rosion.The corrosion rate,P W(mm/y),is presented in Table2[19].Potentiodynamic polarisation curves were measured with metallographic polished specimens in5%NaCl saturated with Mg(OH)2that gave a stable pH of$11,using a three-electrode elec-trochemical cell and a scanning rate of1mV sÀ1.After immersion for1h,the polarisation was started from a potential ofÀ250mV SCE (cathodic)relative to the open circuit potential and was stopped at an anodic potential where the anodic current increased dramati-cally.At least two tests were conducted for each microstructure condition;these confirmed the reproducibility of the polarisation curves.The polarisation curves were used to evaluate the corrosion density,i corr,by Tafel extrapolation of the cathodic branch to the corrosion potential,E corr.The corresponding corrosion rate,P i,is included in Table2.Table2presents i corr,P i,E corr and b c values for GWK103in the as cast(F),solution treated(T4)and aged(T6)conditions,evaluated from polarisation curves measured in5%NaCl saturated with Mg(OH)2.Also listed for comparison are P W,P W/P i,and the relative deviation calculated using Eq.(6).The cathodic Tafel slope,b c,was similar for all conditions,indicating similar electrochemical reac-tions for hydrogen evolution;however,the values did have signif-icant variation larger than experimental scatter,which could indicate that there was more than one hydrogen evolution reac-tion.The values of i corr,and the corresponding corrosion rate,P i, for the different microstructure conditions correlated with the corrosion rates measured from weight loss,P W,but did not agree in magnitude.Moreover the ratio P W/P i was not constant,and the relative deviations were large,indicating that the electrochemical method based on Tafel extrapolation of the cathodic polarisation curve did not provide a good measurement of the corrosion rate, in agreement with Song and Atrens[2].The data of Table2relates to the corrosion rate,P W,measured over3days and P i measured soon after specimen immersion(1h)in the solution.3.3.AZ91in3.5%NaClTable3relates to the study by Candan et al.[20]of the corrosion of AZ91alloys,containing0–1wt%Pb,in3.5%NaCl.The corrosion rate was measured by weight loss for72h immersion in3.5%NaCl, P W,and the corrosion rate,P i,evaluated by Tafel extrapolation from polarisation curves measured with a scan rate of1mV SCE/s after 1h immersion in3.5%NaCl.Specimens were polished to1l m diamond,washed with alcohol and washed with distilled water. There were large differences between the two measures of corro-sion rate.The values of the corrosion rate measured by weight loss,P W, given in Table3,are larger than expect for AZ91in3.5%NaCl asTable2i corr,P i,E corr and b c values for GWK103(Mg–10Gd–3Y–0.4Zr)in the F,T4and T6 conditions,evaluated from polarisation curves measured in5%NaCl saturated with Mg(OH)2after solution equilibration for1h.Also presented are values for P W,P W/P i and e i.P W is the corrosion rate measured from the weight loss for immersion in5% NaCl solution for3days.e i was evaluated using Eq.(6).Material i corr(l A cmÀ2)P i(mm/y)E corr(V SCE)b c(V SCE)P W(mm/y)P W/P ie i(%)GWK103–F50 1.1À1.6700.230 3.0 2.763 GWK103–T4240.54À1.7100.1700.390.7138 GWK103–T6–16h310.71À1.6900.200 2.0 2.865GWK103–T6–500h 300.67À1.6800.190 1.7 2.661Table3Corrosion rates(mm/y)for AZ91base alloys containing0–1%Pb,measured by weightloss,P W,for72h immersion in3.5%NaCl;and P i,evaluated by Tafel extrapolationfrom polarisation curves measured after1h immersion in3.5%NaCl.Alloy P W P i P W/P iAZ91160 3.545AZ91+0.2%Pb110 2.055AZ91+0.5%Pb900.20450AZ91+1.0%Pb800.071140Table1Measurements related to corrosion rate in1M NaCl at room temperature.Hydrogen evolution rate,V H,and weight loss rate,D W,was measured using48h immersion. Tafel extrapolation evaluated i corr from polarisation curves measured using as-polished specimens soon after solution immersion.Also given is the relative deviation for the corrosion rate determined from Tafel extrapolation,e i,compared with the weight loss rate,calculated using Eq.(6).Alloy V H(ml/cm2/d)P H(mm/y)D W(mg/cm2/d)P W(mm/y)i corr(mA/cm2)P i(mm/y)e i(%)P W/P iZE41 5.914 5.7120.09 2.183 5.7 AZ91D 3.17.1 3.0 6.20.040.9185 6.8PureMg 0.44 1.00.430.900.12 2.72000.33Z.Shi et al./Corrosion Science52(2010)579–588581is evident by a comparison of the data of Table 3and those in Tables 1,4and 8.However,the higher values of corrosion rate are plausible because a small amount of Fe (above the tolerance limit)would cause these higher corrosion rates and such Fe contents could easily arise in their alloy production using raw material ingots from a commercial company.It is plausible that the corrosion rate mea-sured over 72h in the immersion tests becomes manifest during the 72h of the test,whereas a significantly lower corrosion rate was present during the first hour of immersion and measured by the corrosion rate,P i ,evaluated by Tafel extrapolation.Table 4relates to the study of Zhou et al.[21]of the corrosion of AZ91alloys,containing Ca,Sb and Bi Pb,in 3.5%NaCl.The corro-sion rate,P W ,was measured by weight loss for 6days immersion in 3.5%NaCl;and the corrosion rate,P i ,evaluated by Tafel extrap-olation from polarisation curves measured with a scan rate of 1mV SCE /s in 3.5%NaCl.Specimens were polished to 6l m diamond,washed with acetone and washed with distilled water.There were large differences between the two measures of corrosion rate.The relative deviations ranged from 30%to 97%.3.4.AZ91in 0.1M NaClTable 5presents corrosion rates [22,23]for AZ91in 0.1M NaCl measured using weight loss for 100h solution immersion in 0.1M NaCl,P W (mm/y);and corrosion rates,P i -10h ,P i -30h and P i -100h ,cal-culated using Eq.(5)in the present work from the values presented in [22,23]of i corr-10h ,i corr-30h and i corr-100h ,estimated in [22,23]by Tafel extrapolation,from polarisation curves measured after solu-tion exposure of 10h,30h and 100h.The authors indicated that ‘‘there appears to be a direct correlation between the weight loss data and the data collected using electrochemical techniques”,although they did not report the actual numerical values of the cor-rosion rates associated with their electrochemical measurements in their papers [22,23].The samples were ground to 1200grit,cleaned,weighted and introduced into the solution.Potentiody-namic polarisation curves were measured at a relatively rapid scan rate of 4mV SHE /s.The agreement between P W and P i -10h ,P i -30h and P i -100h was not good.This is further explored in Table 6,which pre-sents the ratios P W /P i -10h ,P W /P i -30h and P W /P i -100h ;these ratios should be equal to unity for good agreement.There was not good agreement.The experimental description for the weight loss determination [22,23]indicates that there might have been some corrosion prod-ucts on the specimen surface.That might explain the low reported corrosion rates from weight loss,P W .The values for P W for as cast AZ91in Table 5are significantly lower than the values of P W or P H in Tables 1,3and 8and for P W for comparable alloys in Table 4.The authors [22,23]attributed the low corrosion rates,P W ,to the fact that the tests were carried out in a relatively mild solution.The authors recognized that the corrosion rates measured by Tafel extrapolation were typically much greater than the corrosion rates measured by weight loss,P W .They [22,23]attributed the difference between the two techniques to the fact that the corrosion was local-ized.This issue is dealt with in Section 5.2of the discussion.3.5.Secondary AZ91alloysScharfe et al.[24]carried out a large number of corrosion tests on AZ91base alloys with the addition of extra alloying.For the al-loy designated as ‘‘25”(consisting of AZ91+0.5%Cu),they carried out electrochemical tests and hydrogen evolution measurements under nearly similar conditions so that a comparison is possible.Samples were ground to 1200grit and cleaned with ethanol.Polar-isation curves were measured 0.5h after immersion in 5%NaCl,with starting pH 11.The corrosion rate,calculated from the polar-isation curve was reported as 2.19mm/y [24].The corrosion rate was evaluated to be 5.28mm/y from hydrogen evolution data for this alloy in a nearly similar solution (3.5%NaCl,pH 10)between 70and 90h [24].They attributed the difference in corrosion rate to the possibility that the polarisation curve gave a measurement related to the early stages of corrosion whereas the hydrogen evo-lution data related to long-term steady state corrosion.3.6.Mg–6Zn–Mn–(0.5–2)Si–(0–0.2)CaFig.2relates to the study by Lisitsyn et al.[25]on the corrosion of Mg–6Zn–Mn–(0.5–2)Si–(0–0.2)Ca alloys in 3.5%NaCl saturated with Mg(OH)2.The immersion tests were carried out for 72h and the weight loss was converted to a corrosion rate,P W .Also mea-sured was the corrosion rate from polarisation curves at 1h and 4h (labelled as PD test t =1,and PD test 4h).There was some agreement between P W and the corrosion rate measured from the polarisation curves at 4h (PD test 4h)for some alloys,but poor agreement for the other cases.In general,the corrosion rate mea-sured from the immersion tests,P W gave lower values which may be due to the film tendencies of this testing solution.Table 4Corrosion rates (mm/y)for AZ91base alloys,measured by weight loss,P W ,for 6days immersion in 3.5%NaCl;and P i ,evaluated by Tafel extrapolation from polarisation curves measured in 3.5%NaCl.e i was evaluated using Eq.(6).AlloyP W i corr (mA/cm 2)P i e i (%)P W /P i Mg–9Al–0.6Zn–0.2Mn8.60.0110.259734Mg–9Al–0.8Zn–0.2Mn–0.14Ca 8.80.271 6.230 1.4Mg–9Al–0.8Zn–0.2Mn–0.4Sb 19.90.207 4.776 4.2Mg–9Al–0.8Zn–0.2Mn–0.4Sb–1Bi38.00.76217.4552.1Table 5Corrosion rates (mm/y)for AZ91in 0.1M NaCl measured by weight loss over 100h solution immersion,P W ,and,P i -10h ,P i -30h and P i -100h ,evaluated using Eq.(5)from i corr-10h ,i corr-30h and i corr-100h ,estimated by Tafel extrapolation,from polarisation curves measured after solution exposure of 10h,30h and 100h.Alloy100h 10h30h100hP W (mm/y)i corr-10h (mA/cm 2)P i -10h (mm/y)i corr-30h (mA/cm 2)P i -30h (mm/y)i corr-100h (mA/cm 2)P i -100h (mm/y)As cast 0.350.73170.9321 1.0424T40.290.15 3.40.44100.9021T6–10h 0.310.48110.6415 1.0624T6–16h 0.440.08 1.80.8419 2.2351T6–19h0.620.173.90.77184.43101Table 6Ratios P W /P i -10h ,P W /P i -30h and P W /P i -100h from the data of Table 5.Alloy P W /P i -10h P W /P i -30h P W /P i -100h As cast 0.0210.0170.015T40.850.0290.014T6–10h 0.0280.0210.013T6–16h 0.240.0230.0086T6–19h0.160.0340.0061582Z.Shi et al./Corrosion Science 52(2010)579–5883.7.ZE41in 0–1M NaCl,pH 3–11Table 7relates to the work of Zhao etal.[12];they used the same ZE41and the same procedures as in the research summarised in Section 3.1[15].The hydrogen evolution volume was measured as a function of immersion time,for ZE41immersed in 0M,0.1M and 1M NaCl solutions with pH 3,7and 11.The solutions desig-nated as 0M NaCl consisted of distilled water plus zero added NaCl,adjusted to the desired pH value with HCl and NaOH.The evolved hydrogen volume was essentially zero for the 48h immer-sion in 0M NaCl solutions with pH 7and pH 11,which indicated that there was a low corrosion rate for ZE41in neutral or alkaline solutions without chloride ions.For all other solutions,after an incubation period during which there was a low hydrogen evolu-tion rate,there was an increase in hydrogen evolution volume with increasing immersion time.The incubation period and rate of hydrogen evolution depended on the solution.The incubation per-iod decreased and the hydrogen evolution rate increased with increasing chloride ion concentration at each pH and with decreas-ing pH for each chloride ion concentration.The average corrosion rate,P H (mm/y),evaluated over 48h from the average hydrogen evolution rate for cast ZE41immersed in the various NaCl solutions is presented in Table 7.Potentiodynamic polarisation curves were measured for freshly prepared ZE41samples immediately after immersion in 0M,0.1M and 1M NaCl solutions with pH 3,7and 11.The polarisation curves were used to estimate the corrosion current density ,i corr ,at E corr ,by Tafel extrapolation of the cathodic branch and the cor-responding corrosion rates,P io (mm/y),are presented in Table 7.A comparison of these values from Tafel extrapolation with those calculated from the hydrogen evolution data indicated that (i)the corrosion rates were much higher when estimated from the hydrogen evolution rate and (ii)the corrosion rate estimated from the Tafel extrapolation showed the same trends in influence of pH and chloride ion concentration.The reason for the difference may be that different types of corrosion were measured.The corrosion rate from the Tafel extrapolation may relate to the onset of corro-sion,whereas the corrosion rate from the hydrogen evolution mea-surements relates to corrosion averaged over a considerable time period and includes corrosion some considerable time after corro-sion onset,when the corrosion is well established.Polarisation curves were also measured after 48h immersion in some of the solutions.There were large differences compared with the curves measured immediately after solution immersion for each solution.Tafel extrapolation was used to evaluate i corr and the corresponding corrosion rates have been designated,P i-ss ,to indicate that steady state corrosion conditions had been estab-lished.The corrosion rate,P i-ss ,was always greater than the corro-sion rate,P io ,but the difference was small for the 0.1M NaCl solutions whereas the difference was larger in the 1M NaCl solu-tion.The trends for the corrosion rate,P i-ss (related to pH and chlo-ride concentration)were similar to those for the corrosion rate,P H ;but any similarity of the numerical value of P i-ss and P H appears fortuitous.Table 7showed that the corrosion rate determined from the current at the free corrosion potential,did not agree with direct measurements evaluated from the evolved hydrogen.Of most con-cern may be that there did not appear to be any relation between P i-ss and P H .Any similarity of the numerical value of P i-ss and P H ap-pears fortuitous;the ratio P H /P i-ss varied seemingly randomly be-tween 5.0and 1.2.In most cases the relative deviation was large.3.8.Mg alloys in 1M NaClTable 8relates to the work of Zhao et al.[14].The corrosion rates of common Mg alloys (pure Mg,AZ31,AZ91,AM30,AM60,ZE41)immersed in 3wt.%NaCl for 12days were evaluated by mea-suring the hydrogen evolution.The corrosion rate was also esti-mated using Tafel extrapolation of the cathodic branch of the polarisation curve measured soon after specimen immersion in the solution,after 1day and after 7days immersion in the solution.The pure Mg,AZ91,AM60and ZE41were from cast ingot whereas AZ31and AM30were from extrusions.These alloys were high pur-ity and so were suitable for the study of the influences on corrosion other than the effect of the impurity elements.The hydrogen evolution was measured for two samples each of each Mg alloy immersed in 3%NaCl.For pure Mg and the Mg alloys (with the exception of AZ31),there was initially an incubation per-iod during which there was a small rate of hydrogen evolution.The incubation period was quiet long for pure Mg,there was essentially no incubation period for ZE41.Thereafter there was an increase in hydrogen evolution with increasing immersion time.For most al-loys,the rate of hydrogen evolution initially increased with increasing exposure time,which is attributed to corrosion occur-ring over increasing fractions of the surface as was observed in our prior work [12,15].Nevertheless,it should also be noted that the increase in hydrogen evolution could also be associated with the increased actual surface area;the actual surface area of a corroded surface is larger than the original surface area due to anTable 7The corrosion rate (mm/y),P i-ss (estimated from i corr from polarisation curves measured for ZE41after reaching steady state corrosion conditions,i.e.after 48h immersion)compared with the corrosion rate,P io (estimated from i corr measured from polarisation curves for freshly prepared cast ZE41)and the corrosion rate,P H (evaluated over 48h from the average hydrogen evolution rate)for cast ZE41immersed in various NaCl solutions.The units for the corrosion rate were mm/y.(The relative deviation,calculated using Eq.(7)is given in the brackets in each case,%.)NM indicates not measured.pH0.1M NaCl 1M NaCl P HP ioP i-ss P H P ioP i-ss39.7 3.7(62%) 4.5(54%)20 5.0(75%)17.0(4%)7 2.30.63(73%)NM14 1.6(89%) 3.2(77%)111.50.22(85%)0.3(80%)8.00.6(93%)NMFig.2.Corrosion rate data from the study by Lisitsyn et al.[25]on the corrosion of Mg–6Zn–Mn–(0.5–2)Si–(0–0.2)Ca alloys in 3.5%NaCl saturated with Mg(OH)2.The immersion tests were carried out for 72h and the weight loss was converted to a penetration rate,P W .Also measured was the corrosion rate from polarisation curves at 1h and 4h (labelled as PD test t =1,and PD test 4h).There was some agreement between P W and the corrosion rate measured from the polarisation curves at 4h (PD test 4h)for some alloys,but poor agreement for the other cases.In general,the corrosion rate measured from the immersion tests,P W gave lower values which may be due to the film tendencies of this testing solution.Z.Shi et al./Corrosion Science 52(2010)579–588583。

在查文献的时候,经常会发现找不到期刊属于哪个数据库.为了方便大家查阅文献,现发个有用的东东:化学期刊所属数据库列表A:Accounts of Chemical Research 1968- (ACS)Accreditation and Quality Assurance: Journal for Quality, Comparability and Reliability in Chemical Measurement1996- (Springer)Acta Chimica Slovenica 1998-Acta Crystallographica Section A 2000- (Blackwell)Acta Crystallographica Section B 2004- (Blackwell)Acta Crystallographica Section C 2000- (Blackwell)Acta Crystallographica Section D 2000- (Blackwell)Acta Crystallographica Section E 2001- (Blackwell)Acta Crystallographica Section F 2005- (Blackwell)Acta Materialia 1997- (Elsevier)Additives for Polymers 1997- (Elsevier)Adsorption 1998- (Springer-Kluwer)Advanced Drug Delivery Reviews 1997- (Elsevier)Advanced Functional Materials 2001- (Wiley)Advanced Materials 1996- (Wiley)Advanced Synthesis & Catalysis 2001- (Wiley)(Formerly: Journal für Praktische Chemie 1999-2000)Advances in Biochemical Engineering/Biotechnology 1998- (Springer)Advances in Colloid and Interface Science 1995- (Elsevier)Advances in Environmental Research 1997- (Elsevier)Advances in Polymer Science 1997- (Springer)Advances in Polymer T echnology 1996- (Wiley)Aldrichimica Acta 1997-Amino Acids 2000- (Springer)The Analyst 1997- (RSC)The Analyst 1876-1997Analytical Biochemistry 1997- (Elsevier)Analytica Chimica Acta 1995- (Elsevier)Analytical Abstracts 1980- (RSC)Analytical and Bioanalytical Chemistry 2002- (Springer)(formerly Fresenius' Journal of Analytical Chemistry)Analytical Chemistry (Washington, D.C.) 1947- (ACS)I&EC Analytical Edition (1929-1946)Analytical Communications 1997-1999 (RSC)Analytical Communications 1996-1997Analytical Proceedings including Analytical Communications 1994-1995 (RSC)Analytical Proceedings 1980-1993 (RSC)Angewandte Chemie International Edition 1998- (Wiley)Angewandte Chemie International Edition in English 1962 - 1997 (Wiley) Annual Reports on Analytical Atomic Spectroscopy 1971-1984 (RSC)Annual Reports on the Progress of Chemistry 1904-1966 (RSC)Annual Reports on the Progress of Chemistry, Section A, Inorganic Chemistry 1997- (RSC)Annual Reports on the Progress of Chemistry, Section A, Inorganic Chemistry 1979-1997Annual Reports on the Progress of Chemistry, Section A, Physical and Inorganic Chemistry 1973-1978 (RSC)Annual Reports on the Progress of Chemistry, Section A, General Physical and Inorganic Chemistry 1967-1972 (RSC)Annual Reports on the Progress of Chemistry, Section B, Organic Chemistry 1997- (RSC)Annual Reports on the Progress of Chemistry, Section B, Organic Chemistry1967-1997Annual Reports on the Progress of Chemistry, Section C, Physical Chemistry 1997- (RSC)Annual Reports on the Progress of Chemistry, Section C, Physical Chemistry1979-1997Annual Review of Materials Research 1984-Antiviral Research 1997- (Elsevier)Applied Biomaterials 1996- (Wiley)Applied Catalysis A: General 1997- (Elsevier)Applied Catalysis B: Environmental 1997- (Elsevier)Applied Clay Science 1997- (Elsevier)Applied Composite Materials 1997- (Springer-Kluwer)Applied Mathematics and Optimization 1996- (Springer)Applied Microbiology and Biotechnology 1995- (Springer)Applied Organometallic Chemistry 1996- (Wiley)Applied Physics A: Materials Science & Processing 1995- (Springer)Applied Physics B: Lasers and Optics 1996- (Springer)Applied Surface Science 1995- (Elsevier)Archives of Biochemistry and Biophysics 1997- (Elsevier)Archive for History of Exact Sciences 1998- (Springer)Archives of Environmental Contamination and T oxicology 2000- (Springer) Archives of Insect Biochemistry and Physiology 1996- (Wiley)Australian Journal of Chemistry 1997-B:Biochemical and Biophysical Research Communications 1997- (Elsevier) Biochemical Engineering Journal 1997- (Elsevier)Biochemical Pharmacology 1997- (Elsevier)Biochemical Systematics and Ecology 1997- (Elsevier)Biochemistry 1962- (ACS)Biochemistry (Moscow) 2000- (Springer-Kluwer)Biochemistry and Cell Biology 2001- (NRC)Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics 1997- (Elsevier) Biochimica et Biophysica Acta (BBA) - Bioenergetics 1997- (Elsevier) Biochimica et Biophysica Acta (BBA) - Biomembranes 1997- (Elsevier) Biochimica et Biophysica Acta (BBA) - Enzy mology and Biological Oxidation 1997- (Elsevier)Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression 1997- (Elsevier)Biochimica et Biophysica Acta (BBA) - General Subjects 1997- (Elsevier) Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1997- (Elsevier)Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1997- (Elsevier)Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1997- (Elsevier) Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 1997- (Elsevier)Biochimica et Biophysica Acta (BBA) - Reviews on Bioenergetics 1997- (Elsevier)Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1997- (Elsevier) Biochimica et Biophysica Acta (BBA) - Specialized Section on Enzymological Subjects 1997- (Elsevier)Biochimie 1997- (Elsevier)Bioconjugate Chemistry 1990- (ACS)Bioelectrochemistry 2000- (Elsevier)Bioelectrochemistry and Bioenergetics 1995-1999 (Elsevier)Biological & Pharmaceutical Bulletin 1999-Biomacro molecules 2000- (ACS)Biomaterials 1997- (Elsevier)BioMetals 1997- (Springer-Kluwer)Biomedical Chromatography 1997- (Wiley)Bioorganic Chemistry 1995- (Elsevier)Bioorganic & Medicinal Chemistry 1995- (Elsevier)Bioorganic & Medicinal Chemistry Letters 1995- (Elsevier)Biophysical Chemistry 1995- (Elsevier)Biopolymers 1996- (Wiley)Bioprocess and Biosystems Engineering 1996- (Springer)Bioscience Biotechnology and BiochemistryBiosensors and Bioelectronics 1995- (Elsevier)Biotechnology Advances 1997- (Elsevier)Biotechnology and Bioengineering 1996- (Wiley)Biotechnology Letters 1997- (Springer-Kluwer)Biotechnology Progress 1990- (ACS)Biotechnology T echniques 1997-1999 (Springer)Bulgarian Chemical CommunicationsBulletin of the Chemical Society of Japan 1995-Bulletin of Environmental Contamination and T oxicology 2001- (Springer) Bulletin of the Polish Academy of Science, ChemistryC:Canadian Journal of Chemistry 2001- (NRC)Carbohydrate Research 1995- (Elsevier)Catalysis Communications 1997- (Elsevier)Catalysis Letters 1997- (Springer-Kluwer)Catalysis Surveys from Asia 1997- (Springer-Kluwer)Catalysis T oday 1997- (Elsevier)Catalysts & Catalysed Reactions (RSC)CATTECH 2000-2003 (Springer)Cell and Tissue Banking 2000- (Springer)Cellulose 1997- (Springer-Kluwer)Cellulose Chemistry and T echnologyCentral European Journal of Chemistry 2003- (CESJ)ChemBioChem 2000- (Wiley)Chemical Biology V irtual Journal 2002- (RSC)Chemical Co mmunications 1997- (RSC)Chemical Co mmunications 1996-1997Chemical Co mmunications (London) 1965-1968 (RSC)The Chemical Educator 1997- (Springer)Chemical & Engineering News 1998- (ACS)Chemical Engineering Science 1997- (Elsevier)Chemical Engineering & T echnology 1998- (Wiley)Chemical Hazards in Industry 1981- (RSC)Chemical Health and Safety 1995- (Elsevier)Chemical Market Report 1996-Chemical and Petroleum Engineering 2000- (Springer)Chemical & Pharmaceutical Bulletin 1999-Chemical Physics 1995- (Elsevier)Chemical Physics Letters 1995- (Elsevier)The Chemical Record 2001- (Wiley)Chemical Research in Chinese UniversitiesChemical Research in T oxicology 1988- (ACS)Chemical Reviews 1924- (ACS)Chemical Science 2004- (RSC)Chemical Society Reviews 1998- (RSC)Chemical Society Reviews 1972-1997Chemie in unserer Zeit 2000- (Wiley)Chemistry - A European Journal 1998- (Wiley)Chemistry & Biodiversity 2004- (Wiley)Chemistry & Biology 1995- (Elsevier)Chemistry in Britain 2000-2003 (RSC)Chemistry of Heterocyclic Co mpounds 2000- (Springer-Kluwer)Chemistry & IndustryChemistry International 1997-Chemistry Letters 1997-Chemistry of Materials 1989- (ACS)Chemistry of Natural Co mpounds 2000- (Springer-Kluwer)Chemistry and Physics of Lipids 1997- (Elsevier)Chemistry and T echnology of Fuels and Oils 2000- (Springer-Kluwer) Chemistry World 2004- (RSC)Chemo metrics and Intelligent Laboratory Systems 1998- (Elsevier) Chemosphere 1997- (Elsevier)ChemPhysChem 2000- (Wiley)Chemtracts 2000-Chinese Chemical Letters 1999-Chinese Journal of Chemistry 2005- (Wiley)Chinese Journal of Geochemistry 1985-Chinese Journal of Polymer Science 2000- (Springer)Chinese Journal of Reactive PolymersChinese Science BulletinChimia 1997-Chirality 1996- (Wiley)Chromatographia 2003- (Springer)Collection of Czechoslovak Chemical Communications 1994-Colloid Journal 2000- (Springer-Kluwer)Colloid & Polymer Science 1998- (Springer)Colloids and Surfaces A: Physicochemical and Engineering Aspects 1995- (Elsevier)Colloids and Surfaces B: Biointerfaces 1995- (Elsevier)Combinatorial Chemistry - an Online Journal 2003- (Elsevier)Comptes Rendus de l'Academie Bulgare des SciencesComptes Rendus de l'Académie des Sciences - Series IIB -Mechanics-Physics-Chemistry-Astronomy 1995- (Elsevier)Comptes Rendus de l'Académie des Sciences - Series IIC - Chemistry 1998-2001 (Elsevier)Comptes Rendus Chimie 2002- (Elsevier)Computational and Theoretical Polymer Science 1997-2001 (Elsevier) Computational Biology and Chemistry 2003- (Elsevier)Computers & Chemistry 1995-2002 (Elsevier)Computing and V isualization in Science 1997- (Springer)Concepts in Magnetic Resonance Part A 1997- (Wiley)Concepts in Magnetic Resonance Part B: Magnetic Resonance Engineering 2001- (Wiley)Contemporary Organic Synthesis 1994-1997 (RSC)Continuum Mechanics and Thermodynamics 1995- (Springer)Contributions to Mineralogy and Petrology 1995- (Springer)Coordination Chemistry Reviews 1995- (Elsevier)Critical Reviews in Analytical Chemistry 2003- (Elsevier)Croatica Chemica Acta 1996-Crystal Growth & Design 2001- (ACS)Crystal Engineering 1999- (Elsevier)CrystEngComm 1999- (RSC)Current Biology 1997- (Elsevier)Current Medicinal Chemistry 2000- (Bentham) (下载原文与图书馆联系)Current Medicinal Chemistry - Anti-Cancer Agents 2001- (Bentham)(下载原文与图书馆联系)Current Opinion in Biotechnology 1997- (Elsevier)Current Opinion in Cell Biology 1997- (Elsevier)Current Opinion in Chemical Biology 1997- (Elsevier)Current Opinion in Colloid & Interface Science 1999- (Elsevier)Current Opinion in Genetics & Development 1997- (Elsevier)Current Opinion in Pharmacology 1997- (Elsevier)Current Opinion in Plant Biology 1997- (Elsevier)Current Opinion in Structural Biology 1997- (Elsevier)Current Organic Chemistry 2000- (Bentham) (下载原文与图书馆联系)Current T opics in Medicinal Chemistry 2001- (Bentham) (下载原文与图书馆联系) D:Dalton Transactions 1997- (RSC)Discussions of the Faraday Society 1947-1971 (RSC)Doklady Biochemistry and Biophysics 2000- (Springer-Kluwer)Doklady Chemistry 2000- (Springer-Kluwer)Doklady Physical Chemistry 2000- (Springer-Kluwer)Drug Discovery T oday 1997- (Elsevier)Drug of the FutureDyes and Pigments 1997- (Elsevier)E:Electroanalysis 1998- (Wiley)Electrochemistry Communications 1999- (Elsevier)Electrochimica Acta 1995- (Elsevier)Electronic Journal of Theoretical Chemistry 1997 (Wiley) ELECTROPHORESIS 1999- (Wiley)EnantiomerEnergy & Fuels 1987- (ACS)Engineering and ScienceEnvironmental Chemistry Letters 2003- (Springer)Environmental Management 1996- (Springer)Environmental Science & T echnology 1967- (ACS)Environmental T oxicology 1999- (Wiley)Environmental T oxicology and Pharmacology 1997- (Elsevier)Enzyme and Microbial T echnology 1997- (Elsevier)European Food Research and T echnology 1999- (Springer)(formerly Zeitschrift fue Lebensmittel-Untersuchung und -Forschung A) European Journal of Biochemistry 1998- (Blackwell)European Journal of Inorganic Chemistry 1998- (Wiley)European Journal of Medicinal Chemistry 1995- (Elsevier)European Journal of Organic Chemistry 1998- (Wiley)European Journal of Pharmaceutical Sciences 1997- (Elsevier)European Journal of Pharmaceutics and Biopharmaceutics 1997- (Elsevier) European Journal of Pharmacology 1997- (Elsevier)European Journal of Solid State and Inorganic Chemistry 1998 (Elsevier)The European Physical Journal B - Condensed Matter 1998- (Springer)The European Physical Journal D - Atomic, Molecular and Optical Physics 1998- (Springer)The European Physical Journal E - Soft Matter 2000- (Springer)European Polymer Journal 1995- (Elsevier)Experimental Cell Research 1997- (Elsevier)Experimental Neurology 1997- (Elsevier)F:Faraday Discussions 1997- (RSC)Faraday Discussions 1991-1997Faraday Discussions of the Chemical Society 1972-1991 (RSC)Faraday Special Discussions of the Chemical Society (1 issue = N.2) 1972 (RSC) Faraday Sy mposia of the Chemical Society 1972-1984 (RSC)Faraday Transactions 1997-1998 (RSC)FEBS Journal 1995- (Blackwell)FEBS Letters 1997- (Elsevier)FEMS Microbiology Letters 1997- (Elsevier)Fibre Chemistry 2000- (Springer-Kluwer)Fluid Phase Equilibria 1995- (Elsevier)Focus on Polyvinyl Chloride 2002 (Elsevier)Focus on Powder Coatings 2002- (Elsevier)Forensic Science International 1995- (Elsevier)Foundations of Chemistry 1999- (Springer-Kluwer)Free Radical Biology and Medicine 1997- (Elsevier)Fresenius' Journal of Analytical Chemistry 1995-2001 (Springer)Fuel 1995- (Elsevier)Fuel Processing T echnology 1995- (Elsevier)G:Glass and Ceramics 2000- (Springer-Kluwer)Glass Physical and Chemistry 2000- (Springer-Kluwer)Glycobiology 1998-Glycoconjugate Journal 1997- (Springer-Kluwer)Gene 1997- (Elsevier)Geochemical Transactions 2000- (RSC)Granular Matter 1997- (Springer)Green Chemistry 1999- (RSC)H:The Handbook of Environmental Chemistry 2003- (Springer)Helvetica Chimica Acta 1998- (Wiley)Heteroatom Chemistry 1996- (Wiley)Heterocycles 1996-Heterocycles 2000-High Energy Chemistry 2000- (Springer-Kluwer)Holz als Roh- und Werkstoff 1995- (Springer)Hungarian Chemical JournalI:Indian Journal of Chemistry, Sect. AIndian Journal of Chemistry, Sect. BIndustrial & Engineering Chemistry Research 1987- (ACS)I&EC (1909-1970)I&EC Fundamentals (1962-1968)I&EC Process Design and Development (1962-1968)I&EC Product Research and Development (1962-1968)Inorganic Chemistry 1962- (ACS)Inorganic Chemistry Communications 1995- (Elsevier)Inorganic Materials 2000- (Springer-Kluwer)Inorganica Chimica Acta 1995- (Elsevier)Instruments and Experimental T echniques 2000- (Springer-Kluwer) Interface Science 1997- (Springer)International Journal of Adhesion and Adhesives 1997- (Elsevier) International Journal of Antimicrobial Agents 1997- (Elsevier) International Journal of Biochemistry & Cell Biology, The 1997- (Elsevier) International Journal of Biological Macromolecules 1997- (Elsevier) International Journal of Cosmetic Science 1997- (Blackwell) International Journal of Fracture 1997- (Springer-Kluwer)International Journal of Hydrogen Energy 1995- (Elsevier)International Journal of Inorganic Materials 1999-2001 (Elsevier) International Journal of Mass Spectrometry 1998- (Elsevier) International Journal of Mass Spectrometry and Ion Processes 1995-1998 (Elsevier)International Journal of Molecular Sciences 2000-International Journal of Pharmaceutics 1997- (Elsevier)International Journal of Quantum Chemistry 1997- (Wiley)International Journal of Thermophysics 1998- (Springer-Kluwer) International Medicine Journal 2001- (Blackwell)J:Journal of Agricultural and Food Chemistry 1953- (ACS)Journal of Alloys and Compounds 1997- (Elsevier)Journal of the A merican Chemical Society 1879- (ACS)Journal of the A merican College of Cardiology 1997- (Elsevier)Journal of the A merican Society for Mass Spectro metry 1995- (Elsevier) Journal of Analytical and Applied Pyrolysis 1995- (Elsevier)Journal of Analytical Atomic Spectrometry 1997- (RSC)Journal of Analytical Atomic Spectrometry 1986-1997Journal of Analytical Chemistry 2000- (Springer-Kluwer)The Journal of AntibioticsJournal of Applied Crystallography 2000- (Blackwell)Journal of Applied Electrochemistry 1997- (Springer-Kluwer)Journal of Applied Polymer Science 1996- (Wiley)Journal of Applied Spectroscopy 2000- (Springer)Journal of Asian Natural Products ResearchJournal of Basic Microbiology 1998- (Wiley)Journal of Biochemical and Biophysical Methods 1997- (Elsevier)Journal of Biochemical and Molecular T oxicology 1998- (Wiley)Journal of Bioenergetics and Biomembranes 1997- (Springer)Journal of Biological Chemistry 1980-Journal of Biological Inorganic Chemistry 1996- (Springer)Journal of Biological Physics 1997- (Springer)Journal of Bio medical Materials Research Part A 1996- (Wiley)Journal of Bio medical Materials Research Part B: Applied Biomaterials 1996- (Wiley)Journal of Bio molecular NMR 1997- (Springer)Journal of Bioscience and Bioengineering 1997- (Elsevier)Journal of Biotechnology 1997- (Elsevier)Journal of Carbohydrate Chemistry 2001-Journal of Catalysis 1997- (Elsevier)Journal of Cellular Biochemistry 1996- (Wiley)Journal of Chemical Crystallography 1998- (Springer-Kluwer)Journal of Chemical Ecology 1997- (Springer)Journal of Chemical & Engineering Data 1959- (ACS)I&EC Chem and Eng Data Series (1962-1968)Journal of Chemical In formation and Computer Sciences 1975- (ACS) Journal of Chemical Docu mentation (1961-1974)Journal of Chemical Research 1997-1999 (RSC)Journal of the Chemical Society 1862-1877 (RSC)Journal of the Chemical Society (Resumed) 1926-1965 (RSC)Journal of the Chemical Society, Abstracts 1878-1925 (RSC)Journal of the Chemical Society, Chemical Co mmunications 1972-1995 (RSC) Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry1972-1997 (RSC)Journal of the Chemical Society, Faraday Transactions 1990-1997 (RSC) Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases 1972-1989 (RSC)Journal of the Chemical Society, Faraday Transactions 2: Molecular and Chemical Physics 1972-1989 (RSC)Journal of the Chemical Society A: Inorganic, Physical, Theoretical 1966-1971 (RSC)Journal of the Chemical Society B: Physical Organic 1966-1971 (RSC)Journal of the Chemical Society C: Organic 1966-1971 (RSC)Journal of the Chemical Society D: Chemical Co mmunications 1969-1971 (RSC) Journal of the Chemical Society, Perkin Transactions 1 1997-2002 (RSC) Journal of the Chemical Society, Perkin Transactions 1 1972-1997Journal of the Chemical Society, Perkin Transactions 2 1997-2002 (RSC) Journal of the Chemical Society, Perkin Transactions 2 1972-1997Journal of the Chemical Society, Transactions 1878-1925 (RSC)Journal of the Chemical Society of PakistanJournal of Chemical T echnology & Biotechnology 1996- (Wiley)The Journal of Chemical Thermodynamics 1995- (Elsevier)Journal of Chemo metrics 1997- (Wiley)Journal of Chinese Chemical Society (T aipei) 1988-Journal of Chinese Pharmaceutical SciencesJournal of Chro matography A 1995- (Elsevier)Journal of Chro matography B: Analytical T echnologies in the Biomedical and Life Sciences 2002- (Elsevier)(Formerly: Journal of Chro matography B: Biomedical Sciences and Applications 1995-2001)Journal of Chro matography B: Biomedical Sciences and Applications 1995-2001 (Elsevier)Journal of Cluster Science 1997- (Springer-Kluwer)Journal of Colloid and Interface Science 1995- (Elsevier)Journal of Co mbinatorial Chemistry 1999- (ACS)Journal of Co mputational Chemistry 1997- (Wiley)Journal of Co mputer-Aided Materials Design 1997- (Springer-Kluwer)Journal of Co mputer-Aided Molecular Design 1997- (Springer)Journal of Controlled Release 1997- (Elsevier)Journal of Cultural Heritage 1997- (Elsevier)Journal of Elasticity 1997- (Springer-Kluwer)Journal of Electroanalytical Chemistry 1995- (Elsevier)Journal of Electroceramics 1997- (Springer)Journal of Electron Spectroscopy and Related Phenomena 1995- (Elsevier) Journal of Environmental Monitoring 1999- (RSC)Journal of Essential Oil ResearchJournal of Fluorescence 1997- (Springer)Journal of Fluorine Chemistry 1995- (Elsevier)Journal of Food Co mposition and Analysis 1997- (Elsevier)Journal of Food SciencesJournal of Heterocyclic ChemistryJournal of High Resolution Chromatography 1998- (Wiley)Journal of Inclusion Phenomena and Macrocyclic Chemistry 1997- (Springer-Kluwer)Journal of Indian Chemical SocietyJournal of Industrial Microbiology and Biotechnology 1997- (Springer) Journal of Inorganic Biochemistry 1995- (Elsevier)Journal of Inorganic and Organometallic Polymers 1997- (Springer-Kluwer) Journal of Lu minescence 1995- (Elsevier)Journal of Magnetic Resonance 1997- (Elsevier)Journal of Magnetic Resonance, Series A 1995-1996 (Elsevier)Journal of Magnetic Resonance, Series B 1995- (Elsevier)Journal of Mass Spectro metry 1997- (Wiley)Journal of Materials Chemistry 1997- (RSC)Journal of Materials Chemistry 1991-1997Journal of Material Science 1997- (Springer-Kluwer)Journal of Material Science: Materials in Electronics 1997- (Springer-Kluwer) Journal of Material Science: Materials in Medicine 1997- (Springer-Kluwer) Journal of Material Science Letters 1997-2003 (Springer)Journal of Mathematical Chemistry 1997- (Springer-Kluwer)Journal of Medicinal Chemistry 1959- (ACS)Journal of Membrane Science 1997- (Elsevier)Journal of Microbiological Methods 1997- (Elsevier)Journal of Molecular Biology 1997- (Elsevier)Journal of Molecular Catalysis A: Chemical 1997- (Elsevier)Journal of Molecular Catalysis B: Enzymatic 1997- (Elsevier)Journal of Molecular and Cellular Cardiology 1997- (Elsevier)Journal of Molecular Graphics 1995-1996 (Elsevier)Journal of Molecular Graphics and Modelling 1997- (Elsevier)Journal of Molecular Liquids 1995- (Elsevier)Journal of Molecular Modeling 1995- (Springer)Journal of Molecular Recognition 1996- (Wiley)Journal of Molecular Spectroscopy 1995- (Elsevier)Journal of Molecular Structure 1995- (Elsevier)Journal of Molecular Structure: THEOCHEM 1995- (Elsevier)Journal of Nanoparticle Research 1999- (Springer-Kluwer)Journal of Natural Products 1996- (ACS)Journal of Natural Gas ChemistryJournal of Neurochemistry 1994- (Blackwell)Journal of Organic Chemistry 1936- (ACS)Journal of Organometallic Chemistry 1995- (Elsevier)Journal of Peptide Research 1999- (Blackwell)Journal of Peptide Science 1996- (Wiley)Journal of Pharmaceutical and Biomedical Analysis 1995- (Elsevier)Journal of Pharmaceutical Science 1996- (Wiley)Journal of Pharmacological and T oxicological Methods 1997- (Elsevier) Journal of Photochemistry and Photobiology A: Chemistry 1995- (Elsevier) Journal of Photochemistry and Photobiology B: Biology 1995- (Elsevier) Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2000- (Elsevier)The Journal of Physical Chemistry A 1997- (ACS)The Journal of Physical Chemistry B 1997- (ACS)The Journal of Physical Chemistry (1896-1996)Journal of Physical Organic Chemistry 1996- (Wiley)The Journal of Planar Chromatography-Modern TLCJournal of Polymer Research 2002- (Springer-Kluwer)Journal of Polymer Science Part A: Polymer Chemistry 1996- (Wiley) Journal of Polymer Science Part B: Polymer Physics 1996- (Wiley)Journal of Polymers and the Environment 1998- (Springer-Kluwer)Journal of Porous Materials 1997- (Springer-Kluwer)Journal of Power Sources 1995- (Elsevier)Journal and Proceedings of the Institute of Chemistry of Great Britain and Ireland 1920-1943 (RSC)Journal and Proceedings of the Royal Institute of Chemistry 1949(RSC) Journal and Proceedings of the Royal Institute of Chemistry of Great Britain and Ireland 1944-1948 (RSC)Journal für Praktische Chemie 1999-2000 (Wiley)Journal of Proteome Research 2002- (ACS)Journal of Quantitative Spectroscopy and Radiative Transfer 1995- (Elsevier) Journal of Radioanalytical and Nuclear Chemistry 2000- (Springer-Kluwer) Journal of Raman Spectroscopy 1997- (Wiley)Journal of Rare EarthsJournal of the Royal Institute of Chemistry 1950-1964 (RSC)Journal of the Science o f Food and Agriculture 1997- (Wiley)Journal of Separation Science 2001- (Wiley)Journal of Sol-Gel Science and Technology 1997- (Springer-Kluwer)Journal of Solid State Chemistry 1995- (Elsevier)Journal of Solid State Electrochemistry 1997- (Springer)Journal of Solution Chemistry 1997- (Springer-Kluwer)Journal of Structural Biology 1997- (Elsevier)Journal of Structural Chemistry 2000- (Springer-Kluwer)Journal of Structural and Functional Genomics 2000- (Springer)The Journal of Supercritical Fluids 1995- (Elsevier)Journal of Supramolecular Chemistry 2001-2002 (Elsevier)Journal of Synchrotron Radiation 2000- (Blackwell)Journal of Thermal Analysis and Calorimetry 1998- (Springer-Kluwer) Journal of Thermal ScienceJournal of Traditional Chinese MedicineJubilee of the Chemical Society 1891 (RSC)K:Kinetics and Catalysis 2000- (Springer-Kluwer)L:Lab on a Chip 2001- (RSC)Laboratory Automation & Information Management 1998-1999 (Elsevier) Langmuir 1985- (ACS)Laboratory Hazards Bulletin (RSC)Letters in Peptide Science 1997- (Springer)Luminescence 1999- (Wiley)M:Macro molecular Bioscience 2001- (Wiley)Macro molecular Chemistry and Physics 1998- (Wiley)Macro molecular Rapid Communications 1998- (Wiley)Macro molecular Symposia 1999- (Wiley)Macro molecular Theory and Simulations 1998- (Wiley)Macro molecules 1968- (ACS)Magnetic Resonance in Chemistry 1997- (Wiley)Magnetic Resonance in Medicine 1999- (Wiley)Magnetic Resonance Materials in Physics, Biology and Medicine 1995-2002 (Elsevier)Marine Chemistry 1995- (Elsevier)Mass Spectrometry Reviews 1997- (Wiley)Materials Chemistry and Physics 1997- (Elsevier)Materials Letters 1997- (Elsevier)Materials Research Innovations 1996- (Springer)Materials Science 2000- (Springer-Kluwer)Materials Science and Engineering A 1997- (Elsevier)Materials Science and Engineering B 1997- (Elsevier)Materials Science and Engineering C 1995- (Elsevier)Materials Science and Engineering R: Reports 1997- (Elsevier)Materials T oday 1997- (Elsevier)Measurement T echniques 2000- (Springer)Mechanics of Co mposite Materials 2000- (Springer-Kluwer) Mechanics of Ti me-Dependent Materials 1997- (Springer-Kluwer) Medicinal and Aromatic Plants AbstractsMedicinal Research Reviews 1997- (Wiley)Memoirs of the Chemical Society of London 1841 (RSC)Memoirs and Proceedings, Chemical Society, London 1843-1845 (RSC) Mendeleev Communications 1997- (RSC)Methods in Organic Synthesis 2000- (RSC)Mini-Reviews in Medicinal ChemistryMicrochemical Journal 1995- (Elsevier)Microchimica Acta 1999- (Springer)Microfluidics and Nanofluidics 2004- (Springer)Microporous and Mesoporous Materials 1997- (Elsevier)Mineralogy and Petrology 2000- (Springer)Modern Drug Discovery 2000- (ACS)Molecular and Cellular Neuroscience 1997- (Elsevier)Molecular Diversity 1997- (Springer)Molecular Immunology 1997- (Elsevier)Molecular Pharmaceutics 2003- (ACS)Molecular Sieves - Science and T echnology 1998- (Springer) Molecules 1997-Molecules 1997 (Springer)Molecules and Cells 2000 (Springer)Molecules Online 1996-1999 (Springer)Monatshefte fuer Chemie / Chemical Monthly 1998- (Springer)N:Nachrichten aus der ChemieNano Letters 2001- (ACS)Nature 1996-Nature系列月刊:Nature Biotechnology 1999-Nature Cell Biology 1999-Nature Genetics 1999-Nature Immunology 2000-Nature Materials 2002-Nature Medicine 1999-Nature Neuroscience 1998-Nature Structural & Molecular Biology 1999-综述月刊:Nature Reviews Cancer 2001-Nature Reviews Drug Discovery 2002-Nature Reviews Genetics 2000-Nature Reviews Immunology 2001-。

刊名简称刊名全称CHEM REV=L CHEMICAL REVIEWS 化学评论 美国ACCOUNTS CHEM RES ac ACCOUNTS OF CHEMICAL RESEARCH 化学研究述评 美国PROG POLYM SCI pr PROGRESS IN POLYMER SCIENCECHEM SOC REV ch CHEMICAL SOCIETY REVIEWS 化学会评论 英国ALDRICHIM ACTA al ALDRICHIMICA ACTAANNU REV PHYS CHEM an ANNUAL REVIEW OF PHYSICAL CHEMISTRYSURF SCI REP su SURFACE SCIENCE REPORTSSURF SCI REP su SURFACE SCIENCE REPORTSANGEW CHEM INT EDIT an ANGEWANDTE CHEMIE-INTERNATIONAL EDITION 德国应用化学COORDIN CHEM REV co COORDINATION CHEMISTRY REVIEWSNAT PROD REP na NATURAL PRODUCT REPORTSNAT PROD REP na NATURAL PRODUCT REPORTSNAT PROD REP na NATURAL PRODUCT REPORTSADV CATAL ad ADVANCES IN CATALYSISJ AM CHEM SOC jo JOURNAL OF THE AMERICAN CHEMICAL SOCIETY 美国化学会志CATAL REV ca CATALYSIS REVIEWS-SCIENCE AND ENGINEERINGINT REV PHYS CHEM in INTERNATIONAL REVIEWS IN PHYSICAL CHEMISTRYJ PHOTOCH PHOTOBIO C jo JOURNAL OF PHOTOCHEMISTRY AND PHOTOBIOLOGY C-PHOTOCHEM ADV POLYM SCI ad ADVANCES IN POLYMER SCIENCEANAL CHEM an ANALYTICAL CHEMISTRYTOP CURR CHEM to TOPICS IN CURRENT CHEMISTRYTRAC-TREND ANAL CHEM tr TRAC-TRENDS IN ANALYTICAL CHEMISTRYCHEM-EUR J ch CHEMISTRY-A EUROPEAN JOURNAL 化学 德国ADV SYNTH CATAL ad ADVANCED SYNTHESIS & CATALYSISADV SYNTH CATAL ad ADVANCED SYNTHESIS & CATALYSISCHEM COMMUN ch CHEMICAL COMMUNICATIONS 化学通讯 英国ADV ORGANOMET CHEM ad ADVANCES IN ORGANOMETALLIC CHEMISTRYADV ORGANOMET CHEM ad ADVANCES IN ORGANOMETALLIC CHEMISTRY 有机金属化学进展 ORG LETT or ORGANIC LETTERSCURR OPIN COLLOID IN cu CURRENT OPINION IN COLLOID & INTERFACE SCIENCE FARADAY DISCUSS fa FARADAY DISCUSSIONSGREEN CHEM gr GREEN CHEMISTRYSTRUCT BOND st STRUCTURE AND BONDINGSTRUCT BOND st STRUCTURE AND BONDINGADV INORG CHEM ad ADVANCES IN INORGANIC CHEMISTRYCRYST GROWTH DES cr CRYSTAL GROWTH & DESIGNCRYST GROWTH DES cr CRYSTAL GROWTH & DESIGNCRYST GROWTH DES cr CRYSTAL GROWTH & DESIGNCHEM-ASIAN J ch Chemistry-An Asian JournalJ COMPUT CHEM jo JOURNAL OF COMPUTATIONAL CHEMISTRYJ PHYS CHEM B jo JOURNAL OF PHYSICAL CHEMISTRY BJ CHEM THEORY COMPUT jo Journal of Chemical Theory and ComputationADV COLLOID INTERFAC ad ADVANCES IN COLLOID AND INTERFACE SCIENCEINORG CHEM in INORGANIC CHEMISTRYLANGMUIR la LANGMUIR 兰格缪尔 美国BIOMACROMOLECULES bi BIOMACROMOLECULESBIOMACROMOLECULES bi BIOMACROMOLECULESBIOMACROMOLECULES bi BIOMACROMOLECULESJ ORG CHEM jo JOURNAL OF ORGANIC CHEMISTRY 有机化学杂志 美国ORGANOMETALLICS or ORGANOMETALLICSORGANOMETALLICS or ORGANOMETALLICSJ CHROMATOGR A jo JOURNAL OF CHROMATOGRAPHY AJ CHROMATOGR A jo JOURNAL OF CHROMATOGRAPHY AJ ANAL ATOM SPECTROM jo JOURNAL OF ANALYTICAL ATOMIC SPECTROMETRYJ ANAL ATOM SPECTROM jo JOURNAL OF ANALYTICAL ATOMIC SPECTROMETRYJ POLYM SCI POL CHEM jo JOURNAL OF POLYMER SCIENCE PART A-POLYMER CHEMISTRY CRYSTENGCOMM cr CRYSTENGCOMMCRYSTENGCOMM cr CRYSTENGCOMMCHEMPHYSCHEM ch CHEMPHYSCHEMCHEMPHYSCHEM ch CHEMPHYSCHEMANALYST an ANALYSTCURR ORG CHEM cu CURRENT ORGANIC CHEMISTRYPHYS CHEM CHEM PHYS ph PHYSICAL CHEMISTRY CHEMICAL PHYSICSPHYS CHEM CHEM PHYS ph PHYSICAL CHEMISTRY CHEMICAL PHYSICSJ BIOL INORG CHEM jo JOURNAL OF BIOLOGICAL INORGANIC CHEMISTRYJ BIOL INORG CHEM jo JOURNAL OF BIOLOGICAL INORGANIC CHEMISTRYJ PHYS CHEM C jo Journal of Physical Chemistry CJ PHYS CHEM C jo Journal of Physical Chemistry CJ PHYS CHEM C jo Journal of Physical Chemistry CJ AM SOC MASS SPECTR jo JOURNAL OF THE AMERICAN SOCIETY FOR MASS SPECTROMETRY J AM SOC MASS SPECTR jo JOURNAL OF THE AMERICAN SOCIETY FOR MASS SPECTROMETRY J AM SOC MASS SPECTR jo JOURNAL OF THE AMERICAN SOCIETY FOR MASS SPECTROMETRY J CHEM INF MODEL jo Journal of Chemical Information and ModelingJ CHEM INF MODEL jo Journal of Chemical Information and ModelingJ CHEM INF MODEL jo Journal of Chemical Information and Modeling DALTON T da DALTON TRANSACTIONSCHEM REC ch CHEMICAL RECORDORG BIOMOL CHEM or ORGANIC & BIOMOLECULAR CHEMISTRYTALANTA ta TALANTAJ COMB CHEM jo JOURNAL OF COMBINATORIAL CHEMISTRYJ COMB CHEM jo JOURNAL OF COMBINATORIAL CHEMISTRYJ COMB CHEM jo JOURNAL OF COMBINATORIAL CHEMISTRYANAL CHIM ACTA an ANALYTICA CHIMICA ACTAPOLYMER po POLYMERAPPL CATAL A-GEN ap APPLIED CATALYSIS A-GENERALAPPL CATAL A-GEN ap APPLIED CATALYSIS A-GENERALJ MASS SPECTROM jo JOURNAL OF MASS SPECTROMETRYJ MASS SPECTROM jo JOURNAL OF MASS SPECTROMETRYJ MASS SPECTROM jo JOURNAL OF MASS SPECTROMETRYJ PHYS CHEM REF DATA jo JOURNAL OF PHYSICAL AND CHEMICAL REFERENCE DATAJ PHYS CHEM REF DATA jo JOURNAL OF PHYSICAL AND CHEMICAL REFERENCE DATAJ PHYS CHEM REF DATA jo JOURNAL OF PHYSICAL AND CHEMICAL REFERENCE DATAJ PHYS CHEM A jo JOURNAL OF PHYSICAL CHEMISTRY AJ PHYS CHEM A jo JOURNAL OF PHYSICAL CHEMISTRY AANAL BIOANAL CHEM an ANALYTICAL AND BIOANALYTICAL CHEMISTRYANAL BIOANAL CHEM an ANALYTICAL AND BIOANALYTICAL CHEMISTRYMAR CHEM ma MARINE CHEMISTRYMAR CHEM ma MARINE CHEMISTRYEUR J ORG CHEM eu EUROPEAN JOURNAL OF ORGANIC CHEMISTRYTETRAHEDRON te TETRAHEDRONCURR ORG SYNTH cu CURRENT ORGANIC SYNTHESISRAPID COMMUN MASS SP ra RAPID COMMUNICATIONS IN MASS SPECTROMETRYRAPID COMMUN MASS SP ra RAPID COMMUNICATIONS IN MASS SPECTROMETRY ELECTROANAL el ELECTROANALYSISELECTROANAL el ELECTROANALYSISSYNLETT sy SYNLETTNEW J CHEM ne NEW JOURNAL OF CHEMISTRYCRIT REV ANAL CHEM cr CRITICAL REVIEWS IN ANALYTICAL CHEMISTRYCOMMENT INORG CHEM co COMMENTS ON INORGANIC CHEMISTRYMATCH-COMMUN MATH CO ma MATCH-COMMUNICATIONS IN MATHEMATICAL AND IN COMPUTER C MATCH-COMMUN MATH CO ma MATCH-COMMUNICATIONS IN MATHEMATICAL AND IN COMPUTER C MATCH-COMMUN MATH CO ma MATCH-COMMUNICATIONS IN MATHEMATICAL AND IN COMPUTER C J MOL CATAL A-CHEM jo JOURNAL OF MOLECULAR CATALYSIS A-CHEMICALEUR J INORG CHEM eu EUROPEAN JOURNAL OF INORGANIC CHEMISTRYCATAL TODAY ca CATALYSIS TODAYCATAL TODAY ca CATALYSIS TODAYCATAL TODAY ca CATALYSIS TODAYJ SEP SCI jo JOURNAL OF SEPARATION SCIENCETETRAHEDRON-ASYMMETR te TETRAHEDRON-ASYMMETRYTETRAHEDRON-ASYMMETR te TETRAHEDRON-ASYMMETRYTETRAHEDRON-ASYMMETR te TETRAHEDRON-ASYMMETRY 四面体 英国TETRAHEDRON LETT te TETRAHEDRON LETTERS 四面体通讯 英国MICROPOR MESOPOR MAT mi MICROPOROUS AND MESOPOROUS MATERIALSMICROPOR MESOPOR MAT mi MICROPOROUS AND MESOPOROUS MATERIALSMICROPOR MESOPOR MAT mi MICROPOROUS AND MESOPOROUS MATERIALSMICROPOR MESOPOR MAT mi MICROPOROUS AND MESOPOROUS MATERIALSADV PHYS ORG CHEM ad ADVANCES IN PHYSICAL ORGANIC CHEMISTRYADV PHYS ORG CHEM ad ADVANCES IN PHYSICAL ORGANIC CHEMISTRYJ ELECTROANAL CHEM jo JOURNAL OF ELECTROANALYTICAL CHEMISTRYEUR J MED CHEM eu EUROPEAN JOURNAL OF MEDICINAL CHEMISTRYTHEOR CHEM ACC th THEORETICAL CHEMISTRY ACCOUNTSPROG SOLID STATE CH pr PROGRESS IN SOLID STATE CHEMISTRYULTRASON SONOCHEM ul ULTRASONICS SONOCHEMISTRYULTRASON SONOCHEM ul ULTRASONICS SONOCHEMISTRYCATAL COMMUN ca CATALYSIS COMMUNICATIONSSYNTHESIS-STUTTGART sy SYNTHESIS-STUTTGARTJ COLLOID INTERF SCI jo JOURNAL OF COLLOID AND INTERFACE SCIENCETOP CATAL to TOPICS IN CATALYSISTOP CATAL to TOPICS IN CATALYSISCHEM PHYS LETT ch CHEMICAL PHYSICS LETTERSCHEM PHYS LETT ch CHEMICAL PHYSICS LETTERSADV CHROMATOGR ad ADVANCES IN CHROMATOGRAPHYAUST J CHEM au AUSTRALIAN JOURNAL OF CHEMISTRYCOMB CHEM HIGH T SCR co COMBINATORIAL CHEMISTRY & HIGH THROUGHPUT SCREENING COMB CHEM HIGH T SCR co COMBINATORIAL CHEMISTRY & HIGH THROUGHPUT SCREENING COMB CHEM HIGH T SCR co COMBINATORIAL CHEMISTRY & HIGH THROUGHPUT SCREENINGJ FLUORESC jo JOURNAL OF FLUORESCENCEJ FLUORESC jo JOURNAL OF FLUORESCENCEPOLYM DEGRAD STABIL po POLYMER DEGRADATION AND STABILITYEUR POLYM J eu EUROPEAN POLYMER JOURNALPURE APPL CHEM pu PURE AND APPLIED CHEMISTRY 理论化学与应用化学 美国J PHOTOCH PHOTOBIO A jo JOURNAL OF PHOTOCHEMISTRY AND PHOTOBIOLOGY A-CHEMISTRY J ORGANOMET CHEM jo JOURNAL OF ORGANOMETALLIC CHEMISTRYJ ORGANOMET CHEM jo JOURNAL OF ORGANOMETALLIC CHEMISTRYMACROMOL CHEM PHYS ma MACROMOLECULAR CHEMISTRY AND PHYSICSCARBOHYD POLYM ca CARBOHYDRATE POLYMERSCARBOHYD POLYM ca CARBOHYDRATE POLYMERSCARBOHYD POLYM ca CARBOHYDRATE POLYMERSJ SOLID STATE CHEM jo JOURNAL OF SOLID STATE CHEMISTRYJ SOLID STATE CHEM jo JOURNAL OF SOLID STATE CHEMISTRYADV HETEROCYCL CHEM ad ADVANCES IN HETEROCYCLIC CHEMISTRYMICROCHEM J mi MICROCHEMICAL JOURNALCHEM PHYS ch CHEMICAL PHYSICSCHEM PHYS ch CHEMICAL PHYSICSORG PROCESS RES DEV or ORGANIC PROCESS RESEARCH & DEVELOPMENTORG PROCESS RES DEV or ORGANIC PROCESS RESEARCH & DEVELOPMENTCATAL LETT ca CATALYSIS LETTERSINORG CHEM COMMUN in INORGANIC CHEMISTRY COMMUNICATIONSVIB SPECTROSC vi VIBRATIONAL SPECTROSCOPYVIB SPECTROSC vi VIBRATIONAL SPECTROSCOPYVIB SPECTROSC vi VIBRATIONAL SPECTROSCOPYSURF SCI su SURFACE SCIENCESURF SCI su SURFACE SCIENCEJ ANAL APPL PYROL jo JOURNAL OF ANALYTICAL AND APPLIED PYROLYSISJ ANAL APPL PYROL jo JOURNAL OF ANALYTICAL AND APPLIED PYROLYSISUSP KHIM+us USPEKHI KHIMIIPOLYHEDRON po POLYHEDRONPOLYHEDRON po POLYHEDRONCARBOHYD RES ca CARBOHYDRATE RESEARCHCARBOHYD RES ca CARBOHYDRATE RESEARCHCARBOHYD RES ca CARBOHYDRATE RESEARCHSUPRAMOL CHEM su SUPRAMOLECULAR CHEMISTRYINORG CHIM ACTA in INORGANICA CHIMICA ACTAMINI-REV ORG CHEM mi MINI-REVIEWS IN ORGANIC CHEMISTRYSOLID STATE SCI so SOLID STATE SCIENCESSOLID STATE SCI so SOLID STATE SCIENCESSOLID STATE SCI so SOLID STATE SCIENCESCOLLOID SURFACE A co COLLOIDS AND SURFACES A-PHYSICOCHEMICAL AND ENGINEERIN MICROCHIM ACTA mi MICROCHIMICA ACTAADV QUANTUM CHEM ad ADVANCES IN QUANTUM CHEMISTRYJ MOL MODEL jo JOURNAL OF MOLECULAR MODELINGJ MOL MODEL jo JOURNAL OF MOLECULAR MODELINGJ MOL MODEL jo JOURNAL OF MOLECULAR MODELINGJ MOL MODEL jo JOURNAL OF MOLECULAR MODELINGPOLYM INT po POLYMER INTERNATIONALCURR ANAL CHEM cu Current Analytical ChemistryANAL SCI an ANALYTICAL SCIENCESCHEM LETT ch CHEMISTRY LETTERSSEP PURIF REV se SEPARATION AND PURIFICATION REVIEWSSEP PURIF REV se SEPARATION AND PURIFICATION REVIEWSSEP PURIF REV se SEPARATION AND PURIFICATION REVIEWSTHERMOCHIM ACTA th THERMOCHIMICA ACTATHERMOCHIM ACTA th THERMOCHIMICA ACTAJ FLUORINE CHEM jo JOURNAL OF FLUORINE CHEMISTRYJ FLUORINE CHEM jo JOURNAL OF FLUORINE CHEMISTRYCOLLOID POLYM SCI co COLLOID AND POLYMER SCIENCECOLLOID POLYM SCI co COLLOID AND POLYMER SCIENCEJ PHYS ORG CHEM jo JOURNAL OF PHYSICAL ORGANIC CHEMISTRYJ PHYS ORG CHEM jo JOURNAL OF PHYSICAL ORGANIC CHEMISTRYB CHEM SOC JPN bu BULLETIN OF THE CHEMICAL SOCIETY OF JAPAN 日本化学会通J MOL STRUCT jo JOURNAL OF MOLECULAR STRUCTUREJ THERM ANAL CALORIM jo JOURNAL OF THERMAL ANALYSIS AND CALORIMETRYJ THERM ANAL CALORIM jo JOURNAL OF THERMAL ANALYSIS AND CALORIMETRYMAGN RESON CHEM ma MAGNETIC RESONANCE IN CHEMISTRYMAGN RESON CHEM ma MAGNETIC RESONANCE IN CHEMISTRYMAGN RESON CHEM ma MAGNETIC RESONANCE IN CHEMISTRYHELV CHIM ACTA he HELVETICA CHIMICA ACTACALPHAD ca CALPHAD-COMPUTER COUPLING OF PHASE DIAGRAMS AND THERMO CALPHAD ca CALPHAD-COMPUTER COUPLING OF PHASE DIAGRAMS AND THERMO J INORG ORGANOMET P jo JOURNAL OF INORGANIC AND ORGANOMETALLIC POLYMERSJ IRAN CHEM SOC jo Journal of the Iranian Chemical SocietyJ CHEMOMETR jo JOURNAL OF CHEMOMETRICSJ CHEMOMETR jo JOURNAL OF CHEMOMETRICSJ CHEMOMETR jo JOURNAL OF CHEMOMETRICSJ CHEMOMETR jo JOURNAL OF CHEMOMETRICSJ CHEMOMETR jo JOURNAL OF CHEMOMETRICSJ CHEMOMETR jo JOURNAL OF CHEMOMETRICSPOLYM J po POLYMER JOURNALCR CHIM co COMPTES RENDUS CHIMIEJ BRAZIL CHEM SOC jo JOURNAL OF THE BRAZILIAN CHEMICAL SOCIETYINT J QUANTUM CHEM in INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRYINT J QUANTUM CHEM in INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRYINT J QUANTUM CHEM in INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRYSTRUCT CHEM st STRUCTURAL CHEMISTRYSTRUCT CHEM st STRUCTURAL CHEMISTRYSTRUCT CHEM st STRUCTURAL CHEMISTRYSURF INTERFACE ANAL su SURFACE AND INTERFACE ANALYSISAPPL ORGANOMET CHEM ap APPLIED ORGANOMETALLIC CHEMISTRYAPPL ORGANOMET CHEM ap APPLIED ORGANOMETALLIC CHEMISTRYSOLVENT EXTR ION EXC so SOLVENT EXTRACTION AND ION EXCHANGEANAL LETT an ANALYTICAL LETTERSCHROMATOGRAPHIA ch CHROMATOGRAPHIACHROMATOGRAPHIA ch CHROMATOGRAPHIAZ ANORG ALLG CHEM ze ZEITSCHRIFT FUR ANORGANISCHE UND ALLGEMEINE CHEMIE CAN J CHEM ca CANADIAN JOURNAL OF CHEMISTRY-REVUE CANADIENNE DE CHIM CATAL SURV ASIA ca CATALYSIS SURVEYS FROM ASIAMOL SIMULAT mo MOLECULAR SIMULATIONMOL SIMULAT mo MOLECULAR SIMULATIONJ INCL PHENOM MACRO jo JOURNAL OF INCLUSION PHENOMENA AND MACROCYCLIC CHEMIST J INCL PHENOM MACRO jo JOURNAL OF INCLUSION PHENOMENA AND MACROCYCLIC CHEMIST J APPL POLYM SCI jo JOURNAL OF APPLIED POLYMER SCIENCEINT J CHEM KINET in INTERNATIONAL JOURNAL OF CHEMICAL KINETICSJ MATH CHEM jo JOURNAL OF MATHEMATICAL CHEMISTRYJ MATH CHEM jo JOURNAL OF MATHEMATICAL CHEMISTRYARKIVOC ar ARKIVOCJ SOLUTION CHEM jo JOURNAL OF SOLUTION CHEMISTRYB KOR CHEM SOC bu BULLETIN OF THE KOREAN CHEMICAL SOCIETYRADIOCHIM ACTA ra RADIOCHIMICA ACTARADIOCHIM ACTA ra RADIOCHIMICA ACTAJ PORPHYR PHTHALOCYA jo JOURNAL OF PORPHYRINS AND PHTHALOCYANINESMONATSH CHEM mo MONATSHEFTE FUR CHEMIEJ MOL STRUC-THEOCHEM jo JOURNAL OF MOLECULAR STRUCTURE-THEOCHEMJ MOL LIQ jo JOURNAL OF MOLECULAR LIQUIDSJ MOL LIQ jo JOURNAL OF MOLECULAR LIQUIDSJ COAT TECHNOL RES jo Journal of Coatings Technology and ResearchJ PHOTOPOLYM SCI TEC jo JOURNAL OF PHOTOPOLYMER SCIENCE AND TECHNOLOGY HETEROCYCLES he HETEROCYCLESPOLYM BULL po POLYMER BULLETINMOLECULES mo MOLECULESBIOINORG CHEM APPL bi BIOINORGANIC CHEMISTRY AND APPLICATIONSBIOINORG CHEM APPL bi BIOINORGANIC CHEMISTRY AND APPLICATIONSBIOINORG CHEM APPL bi BIOINORGANIC CHEMISTRY AND APPLICATIONS HETEROATOM CHEM he HETEROATOM CHEMISTRYSYNTHETIC COMMUN sy SYNTHETIC COMMUNICATIONSLETT ORG CHEM le LETTERS IN ORGANIC CHEMISTRYJ CHEM SCI jo JOURNAL OF CHEMICAL SCIENCESJ CHROMATOGR SCI jo JOURNAL OF CHROMATOGRAPHIC SCIENCEJ CHROMATOGR SCI jo JOURNAL OF CHROMATOGRAPHIC SCIENCEJ CLUST SCI jo JOURNAL OF CLUSTER SCIENCEJ LIQ CHROMATOGR R T jo JOURNAL OF LIQUID CHROMATOGRAPHY & RELATED TECHNOLOGIE J LIQ CHROMATOGR R T jo JOURNAL OF LIQUID CHROMATOGRAPHY & RELATED TECHNOLOGIECHIMIA ch CHIMIAJPC-J PLANAR CHROMAT jp JPC-JOURNAL OF PLANAR CHROMATOGRAPHY-MODERN TLC TRANSIT METAL CHEM tr TRANSITION METAL CHEMISTRYACTA CHIM SLOV ac ACTA CHIMICA SLOVENICARADIAT PHYS CHEM ra RADIATION PHYSICS AND CHEMISTRYRADIAT PHYS CHEM ra RADIATION PHYSICS AND CHEMISTRYRADIAT PHYS CHEM ra RADIATION PHYSICS AND CHEMISTRYJ CARBOHYD CHEM jo JOURNAL OF CARBOHYDRATE CHEMISTRYJ CARBOHYD CHEM jo JOURNAL OF CARBOHYDRATE CHEMISTRYJ COORD CHEM jo JOURNAL OF COORDINATION CHEMISTRYJ THEOR COMPUT CHEM jo JOURNAL OF THEORETICAL & COMPUTATIONAL CHEMISTRY COLLECT CZECH CHEM C co COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS QUIM NOVA qu QUIMICA NOVAE-POLYMERS e-E-POLYMERSJ POLYM RES jo JOURNAL OF POLYMER RESEARCHJ HETEROCYCLIC CHEM jo JOURNAL OF HETEROCYCLIC CHEMISTRYJ DISPER SCI TECHNOL jo JOURNAL OF DISPERSION SCIENCE AND TECHNOLOGYACTA CHROMATOGR ac ACTA CHROMATOGRAPHICAZ NATURFORSCH B ze ZEITSCHRIFT FUR NATURFORSCHUNG SECTION B-A JOURNAL OF Z NATURFORSCH B ze ZEITSCHRIFT FUR NATURFORSCHUNG SECTION B-A JOURNAL OF INT J MOL SCI in INTERNATIONAL JOURNAL OF MOLECULAR SCIENCESCHINESE J CHEM ch CHINESE JOURNAL OF CHEMISTRYACTA CHIM SINICA ac ACTA CHIMICA SINICAJ MACROMOL SCI A jo JOURNAL OF MACROMOLECULAR SCIENCE-PURE AND APPLIED CHE NAT PROD RES na NATURAL PRODUCT RESEARCHNAT PROD RES na NATURAL PRODUCT RESEARCHHIGH PERFORM POLYM hi HIGH PERFORMANCE POLYMERSPHYS CHEM LIQ ph PHYSICS AND CHEMISTRY OF LIQUIDSPHYS CHEM LIQ ph PHYSICS AND CHEMISTRY OF LIQUIDSORG PREP PROCED INT or ORGANIC PREPARATIONS AND PROCEDURES INTERNATIONAL CROAT CHEM ACTA cr CROATICA CHEMICA ACTACHINESE J ORG CHEM ch CHINESE JOURNAL OF ORGANIC CHEMISTRYPOLYCYCL AROMAT COMP po POLYCYCLIC AROMATIC COMPOUNDSDES MONOMERS POLYM de DESIGNED MONOMERS AND POLYMERSCHINESE J STRUC CHEM ch CHINESE JOURNAL OF STRUCTURAL CHEMISTRYCHINESE J STRUC CHEM ch CHINESE JOURNAL OF STRUCTURAL CHEMISTRYJ ADV OXID TECHNOL jo JOURNAL OF ADVANCED OXIDATION TECHNOLOGIESCENT EUR J CHEM ce CENTRAL EUROPEAN JOURNAL OF CHEMISTRYMENDELEEV COMMUN me MENDELEEV COMMUNICATIONSJ SYN ORG CHEM JPN jo JOURNAL OF SYNTHETIC ORGANIC CHEMISTRY JAPANIRAN POLYM J ir IRANIAN POLYMER JOURNALTURK J CHEM tu TURKISH JOURNAL OF CHEMISTRYTURK J CHEM tu TURKISH JOURNAL OF CHEMISTRYISR J CHEM is ISRAEL JOURNAL OF CHEMISTRYCHEM J CHINESE U ch CHEMICAL JOURNAL OF CHINESE UNIVERSITIES-CHINESEJ CHIN CHEM SOC-TAIP jo JOURNAL OF THE CHINESE CHEMICAL SOCIETYBEILSTEIN J ORG CHEM be Beilstein Journal of Organic ChemistryCHINESE J POLYM SCI ch CHINESE JOURNAL OF POLYMER SCIENCECOLLOID J+co COLLOID JOURNALINDIAN J CHEM A in INDIAN JOURNAL OF CHEMISTRY SECTION A-INORGANIC BIO-IN PHOSPHORUS SULFUR ph PHOSPHORUS SULFUR AND SILICON AND THE RELATED ELEMENTS KINET CATAL+ki KINETICS AND CATALYSISREV ANAL CHEM re REVIEWS IN ANALYTICAL CHEMISTRYACTA PHYS-CHIM SIN ac ACTA PHYSICO-CHIMICA SINICAJ CHEM CRYSTALLOGR jo JOURNAL OF CHEMICAL CRYSTALLOGRAPHYJ CHEM CRYSTALLOGR jo JOURNAL OF CHEMICAL CRYSTALLOGRAPHYANN CHIM-ROME an ANNALI DI CHIMICAANN CHIM-ROME an ANNALI DI CHIMICAINORG REACT MECH in INORGANIC REACTION MECHANISMSINT J POLYM ANAL CH in INTERNATIONAL JOURNAL OF POLYMER ANALYSIS AND CHARACTE SCI CHINA SER B sc SCIENCE IN CHINA SERIES B-CHEMISTRYRES CHEM INTERMEDIAT re RESEARCH ON CHEMICAL INTERMEDIATESJ ANAL CHEM+jo JOURNAL OF ANALYTICAL CHEMISTRYREACT KINET CATAL L re REACTION KINETICS AND CATALYSIS LETTERSCHEM LISTY ch CHEMICKE LISTYCHINESE J INORG CHEM ch CHINESE JOURNAL OF INORGANIC CHEMISTRYJ RADIOANAL NUCL CH jo JOURNAL OF RADIOANALYTICAL AND NUCLEAR CHEMISTRYJ RADIOANAL NUCL CH jo JOURNAL OF RADIOANALYTICAL AND NUCLEAR CHEMISTRYJ RADIOANAL NUCL CH jo JOURNAL OF RADIOANALYTICAL AND NUCLEAR CHEMISTRY CHEM ANAL-WARSAW ch CHEMIA ANALITYCZNAJ CHIL CHEM SOC jo JOURNAL OF THE CHILEAN CHEMICAL SOCIETYPOLYM SCI SER A+po POLYMER SCIENCE SERIES AACTA POLYM SIN ac ACTA POLYMERICA SINICAJ SERB CHEM SOC jo JOURNAL OF THE SERBIAN CHEMICAL SOCIETYIONICS io IONICSIONICS io IONICSIONICS io IONICSRUSS J ORG CHEM+ru RUSSIAN JOURNAL OF ORGANIC CHEMISTRYPROG CHEM pr PROGRESS IN CHEMISTRYHIGH ENERG CHEM+hi HIGH ENERGY CHEMISTRYJ CHEM EDUC jo JOURNAL OF CHEMICAL EDUCATIONJ CHEM EDUC jo JOURNAL OF CHEMICAL EDUCATIONRUSS CHEM B+ru RUSSIAN CHEMICAL BULLETINCHINESE J ANAL CHEM ch CHINESE JOURNAL OF ANALYTICAL CHEMISTRYPOL J CHEM po POLISH JOURNAL OF CHEMISTRYRUSS J COORD CHEM+ru RUSSIAN JOURNAL OF COORDINATION CHEMISTRYCHEM PAP ch CHEMICAL PAPERS-CHEMICKE ZVESTICAN J ANAL SCI SPECT ca CANADIAN JOURNAL OF ANALYTICAL SCIENCES AND SPECTROSCO CAN J ANAL SCI SPECT ca CANADIAN JOURNAL OF ANALYTICAL SCIENCES AND SPECTROSCO STUD CONSERV st STUDIES IN CONSERVATIONSTUD CONSERV st STUDIES IN CONSERVATIONSTUD CONSERV st STUDIES IN CONSERVATIONHETEROCYCL COMMUN he HETEROCYCLIC COMMUNICATIONSREV INORG CHEM re REVIEWS IN INORGANIC CHEMISTRYJ STRUCT CHEM+jo JOURNAL OF STRUCTURAL CHEMISTRYJ STRUCT CHEM+jo JOURNAL OF STRUCTURAL CHEMISTRYLC GC EUR lc LC GC EUROPEPOLYM-KOREA po POLYMER-KOREADOKL PHYS CHEM do DOKLADY PHYSICAL CHEMISTRYCHEM WORLD-UK ch CHEMISTRY WORLDINDIAN J CHEM B in INDIAN JOURNAL OF CHEMISTRY SECTION B-ORGANIC CHEMISTR RUSS J GEN CHEM+ru RUSSIAN JOURNAL OF GENERAL CHEMISTRYCHEM NAT COMPD+ch CHEMISTRY OF NATURAL COMPOUNDSJ RARE EARTH jo JOURNAL OF RARE EARTHSS AFR J CHEM-S-AFR T so SOUTH AFRICAN JOURNAL OF CHEMISTRY-SUID-AFRIKAANSE TYD CHIM OGGI ch CHIMICA OGGI-CHEMISTRY TODAYCHIM OGGI ch CHIMICA OGGI-CHEMISTRY TODAYRUSS J PHYS CHEM A+ru RUSSIAN JOURNAL OF PHYSICAL Chemistry AJ MED PLANTS RES jo Journal of Medicinal Plants ResearchRUSS J INORG CHEM+ru RUSSIAN JOURNAL OF INORGANIC CHEMISTRYCHEM RES CHINESE U ch CHEMICAL RESEARCH IN CHINESE UNIVERSITIESCHINESE CHEM LETT ch CHINESE CHEMICAL LETTERSDOKL CHEM do DOKLADY CHEMISTRYMAIN GROUP MET CHEM ma MAIN GROUP METAL CHEMISTRYMAIN GROUP MET CHEM ma MAIN GROUP METAL CHEMISTRYCHEM UNSERER ZEIT ch CHEMIE IN UNSERER ZEITPROG REACT KINET MEC pr PROGRESS IN REACTION KINETICS AND MECHANISMJ INDIAN CHEM SOC jo JOURNAL OF THE INDIAN CHEMICAL SOCIETYLC GC N AM lc LC GC NORTH AMERICABUNSEKI KAGAKU bu BUNSEKI KAGAKUREV CHIM-BUCHAREST re REVISTA DE CHIMIEREV CHIM-BUCHAREST re REVISTA DE CHIMIEPOLYM SCI SER B+po POLYMER SCIENCE SERIES BINDIAN J HETEROCY CH in INDIAN JOURNAL OF HETEROCYCLIC CHEMISTRYCHEM IND-LONDON ch CHEMISTRY & INDUSTRYOXID COMMUN ox OXIDATION COMMUNICATIONSREV ROUM CHIM re REVUE ROUMAINE DE CHIMIERUSS J APPL CHEM+ru RUSSIAN JOURNAL OF APPLIED CHEMISTRYASIAN J CHEM as ASIAN JOURNAL OF CHEMISTRYB CHEM SOC ETHIOPIA bu BULLETIN OF THE CHEMICAL SOCIETY OF ETHIOPIA AFINIDAD af AFINIDADJ AUTOM METHOD MANAG jo JOURNAL OF AUTOMATED METHODS & MANAGEMENT IN CHEMISTRY J AUTOM METHOD MANAG jo JOURNAL OF AUTOMATED METHODS & MANAGEMENT IN CHEMISTRY KOBUNSHI RONBUNSHU ko KOBUNSHI RONBUNSHUJ CHEM SOC PAKISTAN jo JOURNAL OF THE CHEMICAL SOCIETY OF PAKISTANJ CHEM RES-S jo JOURNAL OF CHEMICAL RESEARCH-SACTUAL CHIMIQUE ac ACTUALITE CHIMIQUERUSS J PHYS CHEM B+ru Russian Journal of Physical Chemistry BCHEM PHYS CARBON ch CHEMISTRY AND PHYSICS OF CARBONCHEM PHYS CARBON ch CHEMISTRY AND PHYSICS OF CARBONCHEM PHYS CARBON ch CHEMISTRY AND PHYSICS OF CARBONJ APPL CRYSTALLOGR jo JOURNAL OF APPLIED CRYSTALLOGRAPHYACTA CRYSTALLOGR B ac ACTA CRYSTALLOGRAPHICA SECTION B-STRUCTURAL SCIENCE ACTA CRYSTALLOGR A ac ACTA CRYSTALLOGRAPHICA SECTION AJ CRYST GROWTH jo JOURNAL OF CRYSTAL GROWTHLIQ CRYST li LIQUID CRYSTALSCRYST RES TECHNOL cr CRYSTAL RESEARCH AND TECHNOLOGYACTA CRYSTALLOGR C ac ACTA CRYSTALLOGRAPHICA SECTION C-CRYSTAL STRUCTURE COM MOL CRYST LIQ CRYST mo MOLECULAR CRYSTALS AND LIQUID CRYSTALSACTA CRYSTALLOGR E ac ACTA CRYSTALLOGRAPHICA SECTION E-STRUCTURE REPORTS ONL CRYSTALLOGR REP+cr CRYSTALLOGRAPHY REPORTSZ KRIST-NEW CRYST ST ze ZEITSCHRIFT FUR KRISTALLOGRAPHIE-NEW CRYSTAL STRUCTURE小类名称(英文)小类分区大类名称大类分区2008年影响因ISSN小类名称(中文0009-2665化学综合CHEMISTRY, MULTIDISCIPLINA1化学123.592 0001-4842化学综合CHEMISTRY, MULTIDISCIPLINA1化学112.176 0079-6700高分子科学POLYMER SCIENCE1化学116.819 0306-0012化学综合CHEMISTRY, MULTIDISCIPLINA1化学117.419 0002-5100有机化学CHEMISTRY, ORGANIC1化学116.733 0066-426X物理化学CHEMISTRY, PHYSICAL1化学114.688 0167-5729物理化学CHEMISTRY, PHYSICAL1化学112.808 0167-5729物理:凝聚态物PHYSICS, CONDENSED MATTER1化学112.808 1433-7851化学综合CHEMISTRY, MULTIDISCIPLINA1化学110.879CHEMISTRY, INORGANIC & NUC1化学110.566 0010-8545无机化学与核化0265-0568医药化学CHEMISTRY, MEDICINAL1化学17.45 0265-0568有机化学CHEMISTRY, ORGANIC1化学17.45 0265-0568生化与分子生物BIOCHEMISTRY & MOLECULAR B2化学17.45 0360-0564物理化学CHEMISTRY, PHYSICAL1化学1 4.812 0002-7863化学综合CHEMISTRY, MULTIDISCIPLINA2化学18.091 0161-4940物理化学CHEMISTRY, PHYSICAL1化学1 5.625 0144-235X物理化学CHEMISTRY, PHYSICAL2化学1 6.892 1389-5567物理化学CHEMISTRY, PHYSICAL2化学1 5.36 0065-3195高分子科学POLYMER SCIENCE1化学2 6.802 0003-2700分析化学CHEMISTRY, ANALYTICAL1化学2 5.712 0340-1022化学综合CHEMISTRY, MULTIDISCIPLINA2化学2 5.27 0165-9936分析化学CHEMISTRY, ANALYTICAL1化学2 5.485 0947-6539化学综合CHEMISTRY, MULTIDISCIPLINA2化学2 5.454 1615-4150应用化学CHEMISTRY, APPLIED1化学2 5.619 1615-4150有机化学CHEMISTRY, ORGANIC1化学2 5.619 1359-7345化学综合CHEMISTRY, MULTIDISCIPLINA2化学2 5.34 0065-3055无机化学与核化CHEMISTRY, INORGANIC & NUC1化学2 3.571 0065-3055有机化学CHEMISTRY, ORGANIC2化学2 3.571 1523-7060有机化学CHEMISTRY, ORGANIC2化学2 5.128 1359-0294物理化学CHEMISTRY, PHYSICAL2化学2 5.493 1364-5498物理化学CHEMISTRY, PHYSICAL2化学2 4.604 1463-9262化学综合CHEMISTRY, MULTIDISCIPLINA2化学2 4.542 0081-5993物理化学CHEMISTRY, PHYSICAL2化学2 6.511 0081-5993无机化学与核化CHEMISTRY, INORGANIC & NUC2化学2 6.511CHEMISTRY, INORGANIC & NUC2化学2 4.214 0898-8838无机化学与核化1528-7483晶体学CRYSTALLOGRAPHY1化学2 4.215MATERIALS SCIENCE, MULTIDI2化学2 4.215 1528-7483材料科学:综合1528-7483化学综合CHEMISTRY, MULTIDISCIPLINA2化学2 4.215 1861-4728化学综合CHEMISTRY, MULTIDISCIPLINA2化学2 4.197 0192-8651化学综合CHEMISTRY, MULTIDISCIPLINA3化学2 3.39 1520-6106物理化学CHEMISTRY, PHYSICAL2化学2 4.189 1549-9618化学综合CHEMISTRY, MULTIDISCIPLINA3化学2 4.274 0001-8686物理化学CHEMISTRY, PHYSICAL3化学2 5.333CHEMISTRY, INORGANIC & NUC2化学2 4.147 0020-1669无机化学与核化0743-7463物理化学CHEMISTRY, PHYSICAL3化学2 4.097 1525-7797高分子科学POLYMER SCIENCE2化学2 4.1461525-7797有机化学CHEMISTRY, ORGANIC2化学2 4.146BIOCHEMISTRY & MOLECULAR B3化学2 4.146 1525-7797生化与分子生物0022-3263有机化学CHEMISTRY, ORGANIC2化学2 3.952CHEMISTRY, INORGANIC & NUC2化学2 3.815 0276-7333无机化学与核化0276-7333有机化学CHEMISTRY, ORGANIC2化学2 3.815 0021-9673分析化学CHEMISTRY, ANALYTICAL2化学2 3.756 0021-9673生化研究方法BIOCHEMICAL RESEARCH METHO2化学2 3.756 0267-9477分析化学CHEMISTRY, ANALYTICAL2化学2 4.028 0267-9477光谱学SPECTROSCOPY2化学2 4.028 0887-624X高分子科学POLYMER SCIENCE2化学2 3.821 1466-8033晶体学CRYSTALLOGRAPHY2化学2 3.535 1466-8033化学综合CHEMISTRY, MULTIDISCIPLINA3化学2 3.535PHYSICS, ATOMIC, MOLECULAR1化学2 3.636 1439-4235物理:原子、分1439-4235物理化学CHEMISTRY, PHYSICAL3化学2 3.636 0003-2654分析化学CHEMISTRY, ANALYTICAL2化学2 3.761 1385-2728有机化学CHEMISTRY, ORGANIC2化学2 3.184PHYSICS, ATOMIC, MOLECULAR2化学2 4.064 1463-9076物理:原子、分1463-9076物理化学CHEMISTRY, PHYSICAL3化学2 4.064CHEMISTRY, INORGANIC & NUC2化学2 3.6 0949-8257无机化学与核化BIOCHEMISTRY & MOLECULAR B3化学2 3.6 0949-8257生化与分子生物MATERIALS SCIENCE, MULTIDI2化学2 3.396 1932-7447材料科学:综合1932-7447物理化学CHEMISTRY, PHYSICAL3化学2 3.396 1932-7447纳米科技NANOSCIENCE & NANOTECHNOLO3化学2 3.396 1044-0305分析化学CHEMISTRY, ANALYTICAL2化学2 3.181 1044-0305光谱学SPECTROSCOPY2化学2 3.181 1044-0305物理化学CHEMISTRY, PHYSICAL3化学2 3.181 1549-9596计算机:信息系COMPUTER SCIENCE, INFORMAT1化学2 3.643COMPUTER SCIENCE, INTERDIS2化学2 3.643 1549-9596计算机:跨学科1549-9596化学综合CHEMISTRY, MULTIDISCIPLINA3化学2 3.643CHEMISTRY, INORGANIC & NUC2化学2 3.58 1477-9226无机化学与核化1527-8999化学综合CHEMISTRY, MULTIDISCIPLINA3化学3 3.477 1477-0520有机化学CHEMISTRY, ORGANIC2化学3 3.55 0039-9140分析化学CHEMISTRY, ANALYTICAL2化学3 3.206 1520-4766应用化学CHEMISTRY, APPLIED1化学3 3.011 1520-4766化学综合CHEMISTRY, MULTIDISCIPLINA3化学3 3.011 1520-4766医药化学CHEMISTRY, MEDICINAL3化学3 3.011 0003-2670分析化学CHEMISTRY, ANALYTICAL2化学3 3.146 0032-3861高分子科学POLYMER SCIENCE2化学3 3.331 0926-860X环境科学ENVIRONMENTAL SCIENCES2化学3 3.19 0926-860X物理化学CHEMISTRY, PHYSICAL3化学3 3.19 1076-5174光谱学SPECTROSCOPY2化学3 2.94 1076-5174生物物理BIOPHYSICS3化学3 2.94 1076-5174有机化学CHEMISTRY, ORGANIC3化学3 2.94 0047-2689物理化学CHEMISTRY, PHYSICAL3化学3 2.424 0047-2689化学综合CHEMISTRY, MULTIDISCIPLINA3化学3 2.424 0047-2689物理:综合PHYSICS, MULTIDISCIPLINARY3化学3 2.424PHYSICS, ATOMIC, MOLECULAR2化学3 2.871 1089-5639物理:原子、分1089-5639物理化学CHEMISTRY, PHYSICAL3化学3 2.871 1618-2642分析化学CHEMISTRY, ANALYTICAL2化学3 3.328 1618-2642生化研究方法BIOCHEMICAL RESEARCH METHO3化学3 3.328 0304-4203海洋学OCEANOGRAPHY2化学3 2.977 0304-4203化学综合CHEMISTRY, MULTIDISCIPLINA3化学3 2.977 1434-193X有机化学CHEMISTRY, ORGANIC3化学3 3.016 0040-4020有机化学CHEMISTRY, ORGANIC3化学3 2.897 1570-1794有机化学CHEMISTRY, ORGANIC3化学3 2.61 0951-4198分析化学CHEMISTRY, ANALYTICAL2化学3 2.772 0951-4198光谱学SPECTROSCOPY3化学3 2.772 1040-0397分析化学CHEMISTRY, ANALYTICAL2化学3 2.901 1040-0397电化学ELECTROCHEMISTRY3化学3 2.901 0936-5214有机化学CHEMISTRY, ORGANIC3化学3 2.659 1144-0546化学综合CHEMISTRY, MULTIDISCIPLINA3化学3 2.942 1040-8347分析化学CHEMISTRY, ANALYTICAL3化学3 3.5CHEMISTRY, INORGANIC & NUC3化学32 0260-3594无机化学与核化MATHEMATICS, INTERDISCIPLI1化学3 3.5 0340-6253数学跨学科应用COMPUTER SCIENCE, INTERDIS2化学3 3.5 0340-6253计算机:跨学科0340-6253化学综合CHEMISTRY, MULTIDISCIPLINA3化学3 3.5 1381-1169物理化学CHEMISTRY, PHYSICAL3化学3 2.814CHEMISTRY, INORGANIC & NUC3化学3 2.694 1434-1948无机化学与核化0920-5861工程:化工ENGINEERING, CHEMICAL1化学3 3.004 0920-5861应用化学CHEMISTRY, APPLIED2化学3 3.004 0920-5861物理化学CHEMISTRY, PHYSICAL3化学3 3.004 1615-9306分析化学CHEMISTRY, ANALYTICAL3化学3 2.746 0957-4166物理化学CHEMISTRY, PHYSICAL3化学3 2.796 0957-4166无机化学与核化CHEMISTRY, INORGANIC & NUC3化学3 2.796 0957-4166有机化学CHEMISTRY, ORGANIC3化学3 2.796 0040-4039有机化学CHEMISTRY, ORGANIC3化学3 2.538MATERIALS SCIENCE, MULTIDI2化学3 2.555 1387-1811材料科学:综合1387-1811应用化学CHEMISTRY, APPLIED2化学3 2.555 1387-1811物理化学CHEMISTRY, PHYSICAL3化学3 2.555 1387-1811纳米科技NANOSCIENCE & NANOTECHNOLO3化学3 2.555 0065-3160物理化学CHEMISTRY, PHYSICAL3化学3 1.833 0065-3160有机化学CHEMISTRY, ORGANIC3化学3 1.833 1572-6657分析化学CHEMISTRY, ANALYTICAL3化学3 2.484 0223-5234医药化学CHEMISTRY, MEDICINAL3化学3 2.882 1432-881X物理化学CHEMISTRY, PHYSICAL3化学3 2.37 0079-6786无机化学与核化CHEMISTRY, INORGANIC & NUC3化学3 2.938 1350-4177声学ACOUSTICS2化学3 2.796 1350-4177化学综合CHEMISTRY, MULTIDISCIPLINA3化学3 2.796 1566-7367物理化学CHEMISTRY, PHYSICAL3化学3 2.791 0039-7881有机化学CHEMISTRY, ORGANIC3化学3 2.47 0021-9797物理化学CHEMISTRY, PHYSICAL3化学3 2.443 1022-5528应用化学CHEMISTRY, APPLIED2化学3 2.212 1022-5528物理化学CHEMISTRY, PHYSICAL3化学3 2.212 0009-2614物理化学CHEMISTRY, PHYSICAL3化学3 2.169。

Colloids and SurfacesA:Physicochemical and Engineering Aspects206(2002)445–454Practical observation of deviation from Lucas–Washburnscaling in porous mediaJoachim Schoelkopf a,b,*,Patrick A.C.Gane a,Cathy J.Ridgway a,G.Peter Matthews ba Omya AG,CH4665Oftringen,Switzerlandb En6ironmental and Fluid Modelling Group,Uni6ersity of Plymouth,Plymouth PL48AA,UKAbstractThis work analyses the applicability of the Lucas–Washburn equation to experimental observations of imbibition into real network structures.The experimental pore structures used in this study are constructed from tablets of two finely ground calcium carbonates,with defined differences in particle size distribution.These are compressed under a range of different applied pressures to achieve a controlled series of porosities while maintaining the surface chemical, particulate and morphological pore characteristics constant.The porosities are determined by mercury intrusion porosimetry applying corrections for mercury compression and penetrometer expansion together with a correction for sample skeletal compression(Gane et al.,J.Am.Chem.Soc.,35(1996)).Imbibition studies are made by bringing each porous sample into contact with a supersource of liquid and the dynamic imbibition is recorded gravimetrically. Results follow a long timescale macroscopic absorption rate depending on the square root of time but show a failure to scale according to pore size in the Lucas–Washburn equation even though the constants of surface energy,contact angle andfluid viscosity have been maintained.Furthermore,values of average measured pore radius are shown to befiner than the Lucas–Washburn predicted equivalent hydraulic capillary radius.The predominance of a relevant pore size within a given pore size distribution forming a selective pathwayfilling based on inertial retardation of larger pores and short-term linear time wetting infiner pores is argued to account for the departure from simple pore size scaling.©2002Elsevier Science B.V.All rights reserved.Keywords:Porous media;Imbibition;Hydraulic radius;Lucas–Washburn;Capillarity;Inertial wetting/locate/colsurfa1.IntroductionThe imbibition of a wetting liquid into a porous structure is a frequently occurring phenomenon in both natural and industrial systems.The stimuli for the work presented here come from the paper and printing industries where imbibition processes are crucially responsible forfinal product quality. More precisely,the imbibition dynamics of an offset ink vehicle,consisting mainly of low viscos-ity mineral oils,into a paper coating structure is the focus of interest.The coating layer is mainly formed byfine mineral pigment particles,today frequently consisting of calcium carbonate.*Corresponding author.Tel.:+41-62-789-2229.E-mail address:joachim.schoelkopf@(J.Schoelkopf).0927-7757/02/$-see front matter©2002Elsevier Science B.V.All rights reserved. PII:S0927-7757(02)00066-3J.Schoelkopf et al./Colloids and Surfaces A:Physicochem.Eng.Aspects206(2002)445–454 446Despite many studies in otherfields of surface chemistry having identified some limitations of the well known Lucas–Washburn equation when applied to the imbibition of liquid into porous substrates[1–5]it is still frequently used,espe-cially in thefield of paper science.In this paper we present new results,using an experimental technique,described earlier[6],which is used to sample directly the mass uptake offluid into a macroscopic consolidated structure offinely ground calcium carbonate.This technique is used to investigate the imbibition of a mineral oil( C16-fraction)into structures over a range of differ-ent porosities,each determined independently by mercury porosimetry adopting the required spe-cially developed compression correction al-gorithms[7].The important aspect of this work is that we maintain the surface chemical and overall geometrical similarity of the samples.Often,these features are either overlooked or,conversely,as-sumed to be changing when correlating the ab-sorption dynamic between different porosity samples in an attempt to support the continued validity of Lucas–Washburn.2.Theoretical backgroundAn early approach to analyse imbibition is reported by Bell and Cameron[16]whofind a root dependency of t for imbibition which was apparently found independently also by Ostwald in1908[11].Lucas experimented with glass tubes andfilter-papers to verify the equation he ob-tained combining the Laplace relation with Poiseuille’s equation of laminarflow[11].His focus was mainly to fortify the square root depen-dency of t.Washburn[12],not being aware of Lucas’work,performing vertical and horizontal capillary experiments,derived the same equation as Lucas and discussed slip behaviour and the limits of Poiseuilleflow at both ends of the liquid column and the equation’s applicability to porous substrates.The main drawback of the combined Lucas–Washburn approach is the lack of inertial terms,relating to the mass offluid under motion, as was recognised by Rideal[17].Bosanquet com-plemented Rideal’s solution in1923[10],adding the inertial impulse drag effect associated with an acceleratingflter,several researchers,for example Szekely et al.[18],being only partially aware of the mentioned classical work,found solutions similar to Bosanquet’s equation adding other(minor)correction terms.In previous studies we investigated the mecha-nism of absorption of polar liquids into coating pigment structures[8,9].We identified the poten-tial relevance of inertialflow as physically pre-dicted by Bosanquet[10],who expanded the well-known Lucas–Washburn[11,12]equation to contain the inertial effect of the liquid mass which has to be accelerated by the wetting force.Ac-cording to the solution of Bosanquet,there exists a time-dependent optimum forflow rate as a function of capillary radius,liquid density and viscosity.The consequence is that pores up to a given diameter in a porous network,this diameter in turn increasing as a function of time,fill very fast while larger features remain by-passed and tend to remain unfilled under conditions of lim-ited supply volumes offluid,as is the case with thinly applied inkfilms or droplets.This promotes a preferential pathwayflow.Both phenomena, inertialflow and unfilled pores,have been ob-served and analysed.Inertialflow in a glass capil-lary,following initially a linear relation with time, was observed directly by Que´re´using a high speed camera[13].The existence of unfilled or by-passed pores is known from soil science and studies made using micro models[14].The experimental method we use provides no direct knowledge about the initial regime of uptake due to the equilibration of the contact forces and experimen-tal resolution over this timescale.Hence it is not possible to determine the expected linear time relation for purely inertial wetting at the time of initial contact.To illustrate these combined issues in relation to a network structure,we previously applied a modified Bosanquet equation to a sequential wet-ting algorithm for Newtonianfluids in a unit cell-based pore space simulator,Pore–Cor1[8].It 1Pore Cor is a software program developed by the Environ-mental and Fluid Modelling Group,University of Plymouth PL48AA,UKJ.Schoelkopf et al./Colloids and Surfaces A:Physicochem.Eng.Aspects206(2002)445–454447could be seen that in each single feature of a porous network,where the liquid becomes accel-erated,inertia acts over a timescale similar to the porefilling time for thefiner pores encountered in paper coatings and leads to a differential in wet-ting front velocity and position during absorption between thefiner and the larger pores.At the present stage,the size of the network simulator unit cell and the computer processing time still limit direct comparison with longer term experi-mentation.In our earlier approach[8],a scaling function was used to extrapolate the initial regime of simulated imbibition of the unit cell where the slope of the uptake curve matches experimental data.Interestingly,on a macroscopic scale a pro-portionality with respect to the square root of time is once again observed due to mass balance criteria.This systematically supports the reason-ing why the long-assumed experimental verifica-tion of Poiseuilleflow,and hence Lucas–Washburn dynamics,has been accepted, with,however,the remaining need for a defined effective capillary radius or surface energy rela-tionship to describe the discrepancy between ab-sorption rates forfine and coarse structures.This issue is discussed further and modelled in some detail in a recent publication by Ridgway and Gane[15].The relevance of considering inertial wetting as a sequential process at the intersection nodes of a network structure containing a range of pore sizes is well-illustrated by this modelling method.In practical applications many recent re-searchers considered the impact of inertia on im-bibition to be negligible due to its relevance only during short time(initial)imbibition.This may be true for imbibition into a long single capillary and for some liquids.Only a few workers in thefield recognised the potential summing effect of inertia in the interconnected void network of a real porous substrate.Sorbie et al.[19]showed the selective mechanism of inertia-retardedflow using a pore-doublet model and by applying Szekely’s equation.High liquid viscosities shift the time of inertialflow into even shorter timescales and low densities decrease the effect of inertial retardation. Even if inertia is manifest,macroscopically a t behaviour may still be observed from a network structure.Thus,concluding that a t behaviour means that inertia plays no role is wrong.In this respect the picture of a single capillary represent-ing a porous substrate is misleading.While in a capillary the inertial regime is relevant only in the initial extremely short time frame of absorption, in a porous network there is an inertial contribu-tion each time the liquid is accelerated.These acceleration events,sometimes observed in the most extreme cases as Haines-jumps,are well-known and observable microscopically.The con-tribution of inertia,therefore,is in terms of a ‘decision-making process’governing which pores fill and which stay only partiallyfilled.The t behaviour shows only that viscosity controls the absorption over the remaining timescale once the decision of which pore willfill is made.Einset[20]compared imbibition rates of differ-ent liquids into particulate structures of carbon and silicon carbide which were characterised by mercury porosimetry.The Lucas–Washburn equation is used to describe the parabolic distance in time,but a discrepancy(1–2orders of magni-tude)was found comparing the obtained pore radii with those of mercury porosimetry.This was explained by an apparent network effect and as-sumed variation of contact angles induced by contaminations.Li et al.[21]used a variety of alkanes for wicking experiments into ceramic structures.They assumed a contact angle of zero, and found an effective radius which was smaller than that afforded by mercury porosimetry by a factor of about2.They questioned the mercury porosimetry result.Our opinion is that mercury porosimetry has shown its reliability with many structures of high bulk moduli and its reliability was confirmed also in our previous work using saturation wetting liquid imbibition methods[6].3.MaterialsWe chose two commercially available dry pow-der products,both derived from natural calcium carbonate,ground under similar chemical condi-tions from the same Orgon,France,limestone source.The grinding is made in a wet state at consistent solids content using a polyacrylate dis-J.Schoelkopf et al./Colloids and Surfaces A:Physicochem.Eng.Aspects206(2002)445–454448Fig.1.Particle size distributions of the coarse andfine CaCO3powders used for consolidation of porous tablets expressed as cumulative ma.%less than an equivalent Stokesian diameter.persing agent applied in proportion to the specific surface area of thefinal pigment size distribution and subsequently dried.To avoid the interference of dispersant molecular weight and size in relation to the pigment size,the two size distributions of the respective products were chosen to be only slightly different in respect to the quantity of particles less than the2m m Stokesian equivalent hydraulic diameter,being90%w/w B2m m and 95%w/w B2m m,defined as‘coarse CaCO3’and ‘fine CaCO3’,respectively.The cumulative particle size distribution curves of the two materials,as measured by sedimentation,2are shown in Fig.1. The bulk samples used in this work are cuboid blocks of each ground pigment,compacted over a range of pressures to form a series of well-defined porosities.The detailed method of powder com-paction,applying pressures up to 260MPa in a steel die on an hydraulic press and subsequent sample grinding,is described elsewhere[6].It proved to give a reproducible and relatively ho-mogeneous porous structure.Such homogeneity is a prequisite so that specimens from the same sample can be used independently for thefluid imbibition and mercury porosimetry experiments. The porosity range achievable with the coarse powder was found to be much broader( 20–40%)than the range achievable with thefine pigment( 26–33%),indicating differences in packing characteristics.The samples,being con-solidated and maintaining their integrity,did not require a sample vessel for thefluid imbibition experiments,thus eliminating uncertainties of fluid interactions between the sample and such a vessel.To reduce artefacts caused by the wetting of their outer surfaces,the samples were coated with a thin barrier line of a silicone polymer3around the base of the vertical edges arising from the basal plane.The remainder of the outer planes were not coated to allow for the free movement of displaced air during liquid imbibition,and to minimise any interaction between the silicone and the absorbed liquid.The liquid used was a mineral oil4(aromatic free quality),as employed typically in the formu-lation of offset printing inks.The contact angle of oil/calcium carbonate,q,was assumed to be zero following the data of Chibowski et al.[22],who have shown that aliphatic alkanes completely wet a number of mineral surfaces including calcium carbonate—this being the basis upon which these authors used alkanes to determine an effective pore radius.Additionally,this wetting behaviour was also confirmed by observing the complete spreading of an oil droplet on a dispersant pre-ad-sorbed macro-crystal surface of calcium carbon-3Dow Corning P4-3117conformal coating.4Haltermann PKWF6/9af.2Measurements made on a Sedigraph5100.J .Schoelkopf et al ./Colloids and Surfaces A :Physicochem .Eng .Aspects 206(2002)445–454449ate.An oil viscosity of 4.3mPa was determined with a StressTech rheometer performing a small ramp of shear rates showing Newtonian be-haviour.The surface tension was measured to be 27.4m Nm −1by the means of a Kru ¨ss Digital Tensiometer K10T.The density of 0.805g cm −3was given by the manufacturer.These values indi-cate that it can be assumed to be an alkane isomeric blend of around C 16.4.Mercury porosimetryEach structure used for the experimentation was analysed independently with mercury porosimetry.A Micromeritics Autopore III mer-cury porosimeter was used to measure the intru-sion characteristics of the samples.The maximum applied pressure of mercury was 414MPa (60000psia),equivalent to a Laplace throat diameter of 0.004m m.Small samples were used,each of around 1.5g in weight.The equilibration time at each of the increasing applied pressures of mer-cury was set to 60s.The mercury intrusion mea-surements were corrected for the compression of mercury,expansion of the glass sample chamber or ‘penetrometer ’and compressibility of the solid phase of the sample by use of the following equation from Gane et al.[7],as incorporated in the software Pore –Comp.5V int =V obs −l V blank+0.175(V I bulk )log 101+P 1820n−VIbulk(1−F I)1−exp(P I −P )M ssn(1)where V int is the volume of intrusion into the sample,V obs the intruded mercury volume read-ing,l V blank the change in the blank run volume reading,V I bulk the sample bulk volume at atmo-spheric pressure,P the applied pressure,F I the porosity at atmospheric pressure,P I the atmo-spheric pressure and M ss the bulk modulus of thesolid sample.The volume of mercury intruded at the maximum pressure,once corrected for sample compression effects,can be used to calculate the porosity of the sample.For convenience,a repre-sentative pore radius is de fined as r 50=d 50/2,where d 50is the pore diameter,at which 50%of the corrected mercury intrusion volume occurs,(Fig.2).The derivation and validation of the d 50diameter is described in many previous publica-tions and represents a well-accepted number based on the observed distribution function of our pore features.These show in the speci fic cases here that intrusion volume as a function of pore-size is a linear function of applied intrusion pres-sure with similar gradients between samples (Fig.2).Since these functions are of the same form we can ignore parameters of the breadth of pore size distribution and use only a single parameter,d 50,to describe them.If a volume other than the 50%filling would have been chosen,the resulting d y %value would be only slightly shifted in either direction and not change the interdependent find-ings reported later.5.Liquid imbibition methodologyThe recording of the position of the liquid front within the sample by eye or camera is imprecise due to the fuzzy appearance of the wetting front.This is assumed to be due to the previously dis-cussed suspected preferred pathway flow at theFig.2.Typical Hg-intrusion curve as a function of the Laplace pressure related pore diameter,with the determined d 50pore value.5Pore-Comp is a software program of the Environmental and Fluid Modelling Group,University of Plymouth PL48AA,UK.J .Schoelkopf et al ./Colloids and Surfaces A :Physicochem .Eng .Aspects 206(2002)445–454450Fig.3.Gravimetric wetting apparatus.Preliminary trials showed that the silicone ring around the basal edge is ef ficient in preventing fluid from creeping up the outside of the sample,so that,to a good approximation,F side =0.F contact ,(see Eq.(3)),caused by the force of attrac-tion around the perimeter of the meniscus pulling the liquid up towards the fixed solid,is constant for t \t 1,which,in the case of the viscous ink,can be signi ficant.F base is caused by the formation of the meniscus and the subsequent movement of fluid through the meniscus;the first effect is com-pleted also in time t 1,and the second is assumed negligible because the meniscus is thin and the curvature slight compared with the total cross-sec-tional area of uptake.There is also inertia in the system which causes a lag and then an overshoot in the recorded weight.This effect is assumed to be completed in a time t 2,which is greater than t 1.Thus,to a good approximation in our experimentation,F total (t \t 2)=F wetting (t \t 2)=F wi (t \t 2)+F base (t \t 2)+F contact (t \t 2)+F side (t \t 2)=F wi (t \t 2)+c(3)The constant term c can be found by fitting the function F total (t \t 2)with a linear regression as a function of the square root of time,and extrapo-lating back to t =0,at which point F wi =0.Then the constant term can be subtracted from F total ,and F wi ,the wicking force or internal wetting force,can be calculated at all times.In practice,the forces are measured as apparent changes in liquid weight.Experiments with five similar sam-ples were shown to have a repeatability within 90.96%in imbibition mass at 1000s [8].6.Analysis of absorption ratesUniquely,we investigate structures over a range of different porosities using two closely related skeletal size distributions forming a range of mean pore radii whilst maintaining the chemical and overall geometrical similarity of the samples.The task is to analyse whether Lucas –Washburn re-liquid front.Therefore,the rate of liquid mass uptake was measured instead using an automated microbalance,namely a PC-linked Mettler Toledo AT460balance with a precision of 0.1mg,capa-ble of 2.7measurements s −1.A software program was developed [8]and improved further in the present work,interfacing with the balance for data sampling.6To provide a suf ficiently slow and precise approach of the sample down to the liquid surface,a special sample holder was constructed (Fig.3)—details of which are given elsewhere [8].All experimentation in this study was maintained under constant temperature conditions of 23.091.5°C.As previously de fined [8],the total force F total acting on the solid –liquid interface during the imbibition of oil into the calcium carbonate net-work structure is the sum of the wetting,gravity and buoyancy forces,all of which are functions of time,t :F total (t )=F wetting (t )+F gravity (t )+F bouyancy (t )(2)6Software available from the authors.J.Schoelkopf et al./Colloids and Surfaces A:Physicochem.Eng.Aspects206(2002)445–454451ally applies for porous network structures with surface properties held constant.All parameters in the Lucas–Washburn equation are known in this experimentation as they have been deter-mined using independent methods.This gives us the opportunity to analyse the network contribu-tion to the apparent pore-radius as if it would satisfy the Lucas–Washburn equation and then to see if this correlates with observation.We begin by defining an equivalent hydraulic radius,r ehc,for an equivalent hydraulic capillary (ehc)which behaves in the same absorptive way as the structures,independently of any precon-ceived absorption theory,related to volume up-take rates.It is also possible to define an ehc based on apparent Darcy imbibition distances or observed liquid front position into the structures as a function of time.The two definitions are not in themselves totally compatible if for any reason the complete porosity of the sample is not beingfilled at a given time,t,behind the wetting front.In a previous paper[23],we used the definition based on Darcy length where a difference of a factor greater than4was found between the measured r50and the derived r ehc for similar compressed tablets to those used in this work.To understand the subtle differences a net-work makes when comparing between the Darcy definition and that defined by volume,let the porous structure that is actuallyfilling be de-scribedfirst as a simple bundle of capillaries. This is only for convenience to visualise the rela-tion between structure capillaries and the ehc and assumes nothing of the mechanism of a net-work structure.The real distance of the wetting front,x,absorbing into this bundle of capillaries is a function of surface energy comparison be-tween thefluid and the skeletal surface;defined by k LV cos q,where k LV is thefluid surface ten-sion between the meniscus and the vapour phase (air)and q describes the contact angle between the advancing meniscus and the solid phase. Defining p as thefluid viscosity and r c as the individual capillary radius in the bundle,and t is the time,thenx=f(k LV cos q,p,r c,t)(4)is an expression for the distance travelled in that capillary at time,t.The volume,V(t),offluid absorbed per unit cross-sectional area,A,of the sample at a given time,t,is therefore equated between the capil-lary bundle and the ehc,V(t)/A=%Ni=1x i(t)y r ci2=x(t)y r ehc2(5) where N is the number of capillaries per unit area accessing the surface of the sample.The Darcy length,x Darcy,is defined asx Darcy=V(t)A(6) where is the measured porosity of the sample. Now,suppose all the capillaries have the same radius,the actual wetting front distance x in the bundle will be the same as in just one capillary. The value of x Darcy,however,is dependent on the sum of the volumes having entered into each capillary,i.e.the number of capillaries and the porosity these represent.Clearly,the porosity of a single capillary per unit area is far less than that of a bundle of N capillaries per unit area, hence the correction using the measured porosity takes care of this in a case where all the porous structure is simultaneously beingfilled behind the wetting front.When we have a distribution of capillary radii,the Darcy length now repre-sents the distance of the wetting front based on the geometric mean of the capillary radii. Consider a complicated system of tortuous paths intertwined,but not interconnected,still giving the same measured porosity.Now again, Darcy length will not relate to the observed wet-ting front,i.e.how far the liquid has imbibed into a complex sample,but x Darcy/x observed repre-sents a tortuosity.Therefore,the concept of Darcy length should not be confused with the progress observed in imbibition into real net-work structures,as it represents the length that would be present if the sample were modelled by a bundled distribution of straight capillaries given by the function of the geometric mean of their radii.If we were to derive an ehc based on Darcy length,as defined by Lucas–Washburn,it would followJ .Schoelkopf et al ./Colloids and Surfaces A :Physicochem .Eng .Aspects 206(2002)445–454452r ehc Darcy =V (t )A22pk LV cos q t= d(V (t )/A ) d22p k LV cos q(7)Themost important drawback using anr ehc Darcy ,(which includes an assumed porosity term),is,therefore,that it pre-supposes the com-plete filling of the available structure from the super source up to the liquid front.We now,therefore,return to de fining an ehc based on experimental volume uptake.As we described above,the measurement from experi-ment is that of liquid mass uptake as a function of time into the porous compressed pigment tablets of de fined porosities.The pigments we know have constant k LV cos q ,and the fluid properties are the same in all experiments.The experimental parameters are therefore:m (t )=V (t )z(8)where m (t )is the mass uptake at time t ,as de fined by a volume V (t )of fluid of density z .We normalise to the cross-sectional area of the sam-ple,A ,such that our data become V (t )/A ,the volume absorbed per unit cross-sectional area of the sample.It is shown experimentally that the rate of volume uptake,as mentioned before,does indeed approximate to a t relationship,(Fig.4).There-fore,as our interest is primarily in rates of uptake,Fig.5.Both structure series,differing in pore size distribution only show similar volume rate imbibition as a function of porosity.we can express each curve as a linear relationship between V (t )/A and t ,the gradient of which we can write as d(V (t )/A )d t=d((m (t )/A )/z )d t(9)and which can be obtained directly from the plotted data by a linear regression analysis.Experimentally,we see that the gradient of uptake volume as a function of t follows a linear relationship with porosity and this describes typically the absorption dynamic of our samples,both coarse and fine,(Fig.5).Assuming firstly the universality of the Lucas –Washburn equation,the volume uptake per unit cross-sectional area of the sample should be ex-pressed in terms of the basic interactional parame-ters between fluid and the solid surface making up the boundaries of the pores as V LW (t )/A =1A y r ehc2'r ehc k LV cos q t2p(10)formed by balancing the Laplace pressure across a curved meniscus with the Poiseuille resistive lami-nar flow in the circular capillary,and letting the volume uptake per unit area equal the volume filled into our equivalent capillary which repre-sents that unit area.This de finition no longer relates directly to the porosity of the sample and the incompatibility with a Darcy ehc ,discussed previously,becomes a natural consequence.We derive from Eq.(10)the r ehc for each struc-ture by comparing the experimental uptake gradi-ents with the assumed parameters of theFig.4.Imbibition curves for increasing porosities (from bot-tom to top,same order as in legend)normalised to [V (t =0)/A ]=0to remove wetting jump.Data shown are for the coarse CaCO 3series.。